

Physiological genomics identifies genetic modifiers of long QT syndrome type 2 severity
- PMID: 29431731
- PMCID: PMC5824853
- DOI: 10.1172/JCI94996
Physiological genomics identifies genetic modifiers of long QT syndrome type 2 severity
Abstract
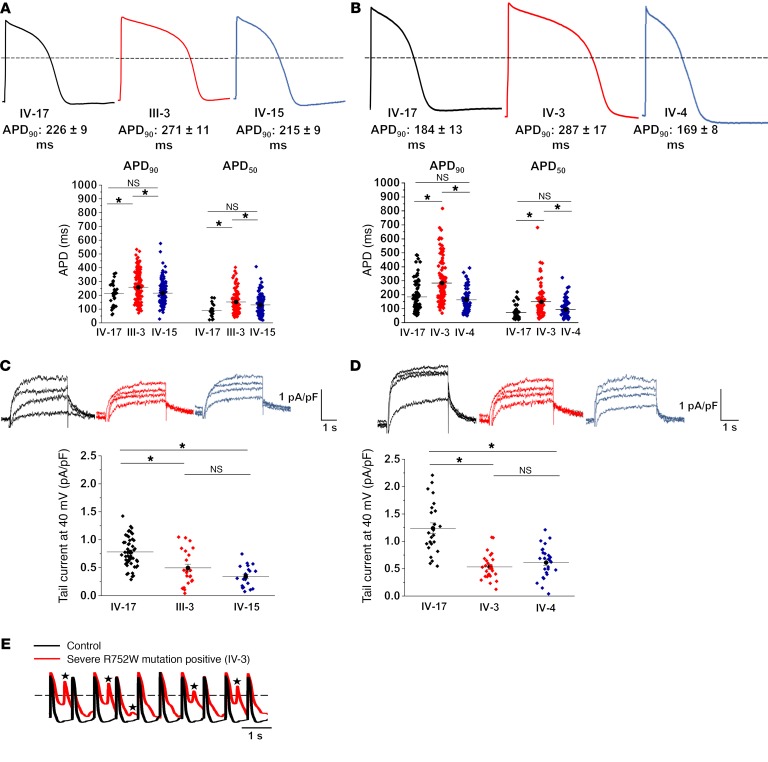
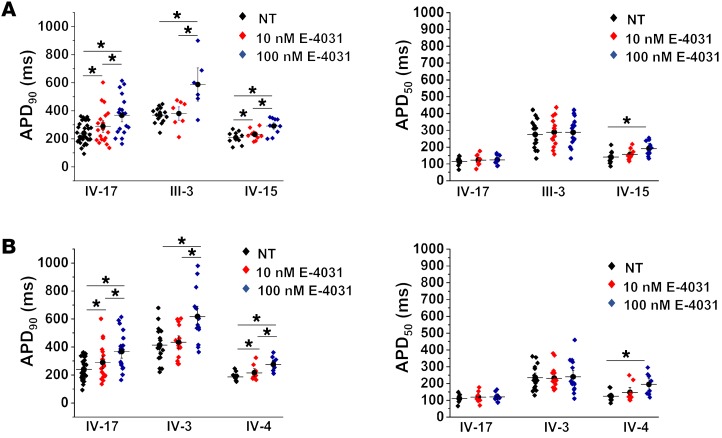
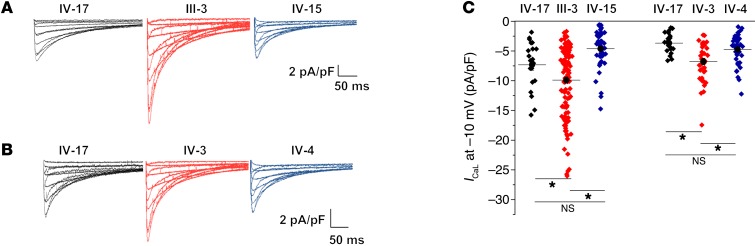
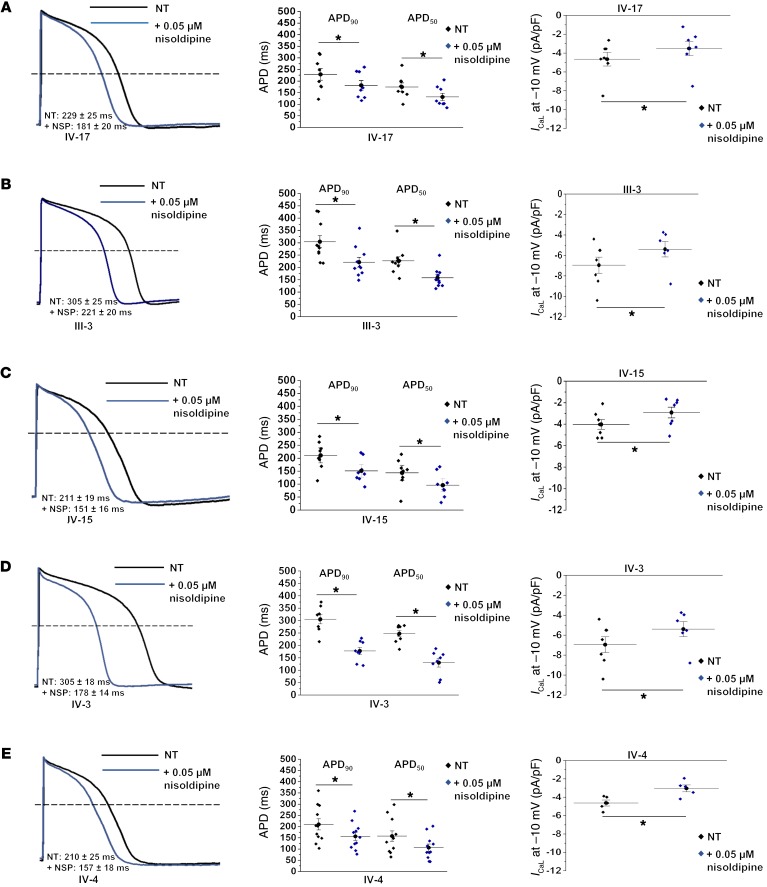
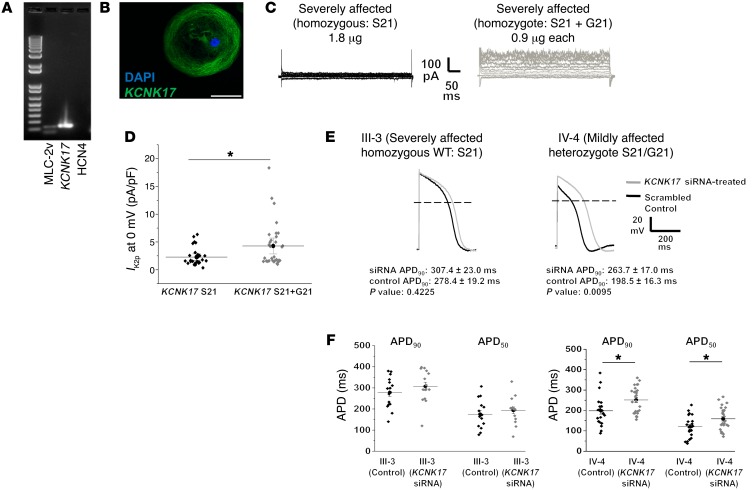
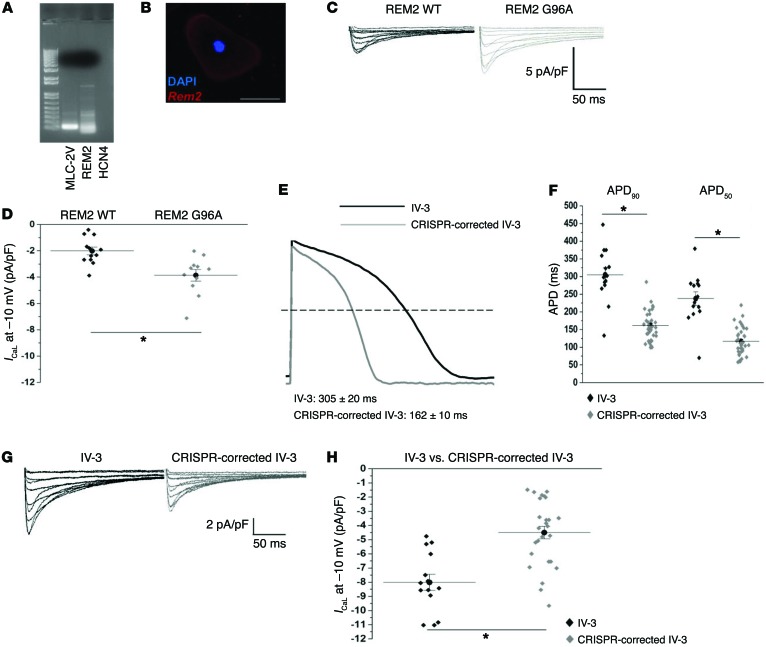
Congenital long QT syndrome (LQTS) is an inherited channelopathy associated with life-threatening arrhythmias. LQTS type 2 (LQT2) is caused by mutations in KCNH2, which encodes the potassium channel hERG. We hypothesized that modifier genes are partly responsible for the variable phenotype severity observed in some LQT2 families. Here, we identified contributors to variable expressivity in an LQT2 family by using induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) and whole exome sequencing in a synergistic manner. We found that iPSC-CMs recapitulated the clinical genotype-phenotype discordance in vitro. Importantly, iPSC-CMs derived from the severely affected LQT2 patients displayed prolonged action potentials compared with cells from mildly affected first-degree relatives. The iPSC-CMs derived from all patients with hERG R752W mutation displayed lower IKr amplitude. Interestingly, iPSC-CMs from severely affected mutation-positive individuals exhibited greater L-type Ca2+ current. Whole exome sequencing identified variants of KCNK17 and the GTP-binding protein REM2, providing biologically plausible explanations for this variable expressivity. Genome editing to correct a REM2 variant reversed the enhanced L-type Ca2+ current and prolonged action potential observed in iPSC-CMs from severely affected individuals. Thus, our findings showcase the power of combining complementary physiological and genomic analyses to identify genetic modifiers and potential therapeutic targets of a monogenic disorder. Furthermore, we propose that this strategy can be deployed to unravel myriad confounding pathologies displaying variable expressivity.
Keywords: Arrhythmias; Cardiology; Genetics; Ion channels; iPS cells.
Conflict of interest statement
Figures

References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
