

Nanopore sequencing and assembly of a human genome with ultra-long reads
- PMID: 29431738
- PMCID: PMC5889714
- DOI: 10.1038/nbt.4060
Nanopore sequencing and assembly of a human genome with ultra-long reads
Abstract
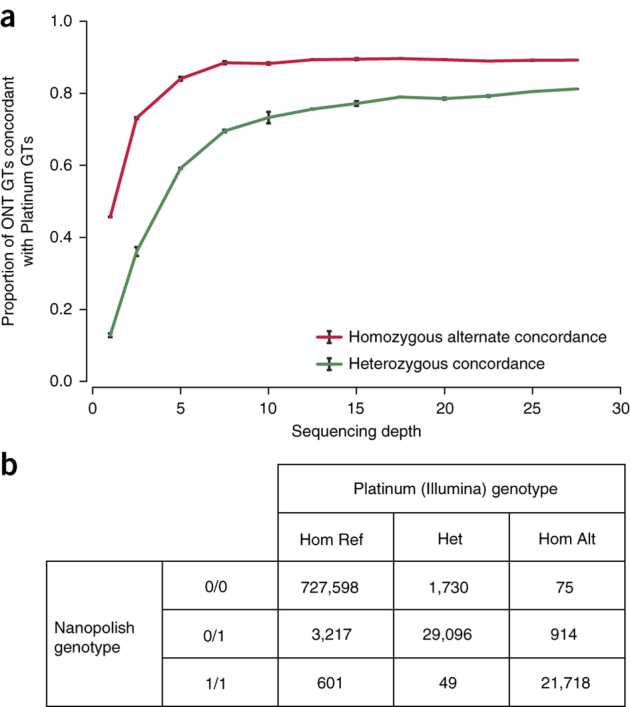
We report the sequencing and assembly of a reference genome for the human GM12878 Utah/Ceph cell line using the MinION (Oxford Nanopore Technologies) nanopore sequencer. 91.2 Gb of sequence data, representing ∼30× theoretical coverage, were produced. Reference-based alignment enabled detection of large structural variants and epigenetic modifications. De novo assembly of nanopore reads alone yielded a contiguous assembly (NG50 ∼3 Mb). We developed a protocol to generate ultra-long reads (N50 > 100 kb, read lengths up to 882 kb). Incorporating an additional 5× coverage of these ultra-long reads more than doubled the assembly contiguity (NG50 ∼6.4 Mb). The final assembled genome was 2,867 million bases in size, covering 85.8% of the reference. Assembly accuracy, after incorporating complementary short-read sequencing data, exceeded 99.8%. Ultra-long reads enabled assembly and phasing of the 4-Mb major histocompatibility complex (MHC) locus in its entirety, measurement of telomere repeat length, and closure of gaps in the reference human genome assembly GRCh38.
Conflict of interest statement
M.L., N.L., J.O.G., J.T.S., J.R.T., and T.P.S. were members of the MinION access program (MAP) and have received free-of-charge flow cells and kits for nanopore sequencing for this and other studies, and travel and accommodation expenses to speak at Oxford Nanopore Technologies conferences. N.J.L. has received an honorarium to speak at an Oxford Nanopore company meeting. S.K., A.T.D., J.Q., and T.A.S. have received travel and accommodation expenses to speak at Oxford Nanopore Technologies conferences. J.T.S., J.O.G., and M.L. receive research funding from Oxford Nanopore Technologies.
Figures




Comment in
-
Errors in long-read assemblies can critically affect protein prediction.Nat Biotechnol. 2019 Feb;37(2):124-126. doi: 10.1038/s41587-018-0004-z. Nat Biotechnol. 2019. PMID: 30670796 No abstract available.
References
Publication types
MeSH terms
Grants and funding
- MR/L015080/1/MRC_/Medical Research Council/United Kingdom
- MR/M009157/1/MRC_/Medical Research Council/United Kingdom
- U41 HG007234/HG/NHGRI NIH HHS/United States
- MR/M501621/1/MRC_/Medical Research Council/United Kingdom
- MR/N013956/1/MRC_/Medical Research Council/United Kingdom
- U24 CA209999/CA/NCI NIH HHS/United States
- BB/M020061/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 102732/WT_/Wellcome Trust/United Kingdom
- BB/N017099/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/R000492/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- T32 GM007464/GM/NIGMS NIH HHS/United States
- U54 HG007990/HG/NHGRI NIH HHS/United States
- R01 HG007827/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
