

Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival
- PMID: 29434213
- PMCID: PMC5833690
- DOI: 10.1038/s41419-017-0242-x
Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival
Abstract
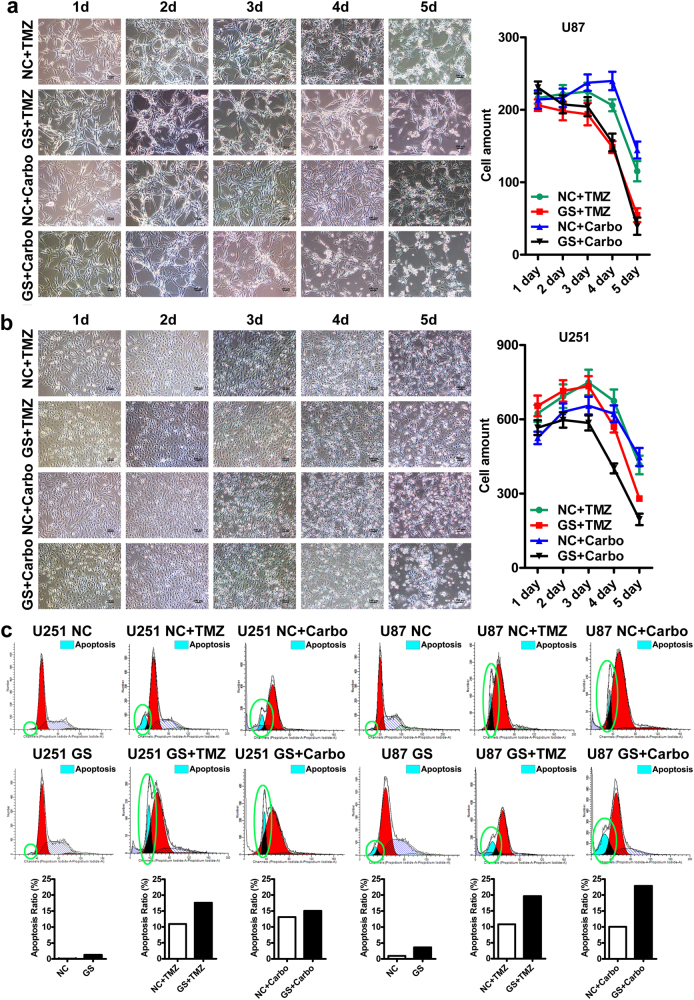
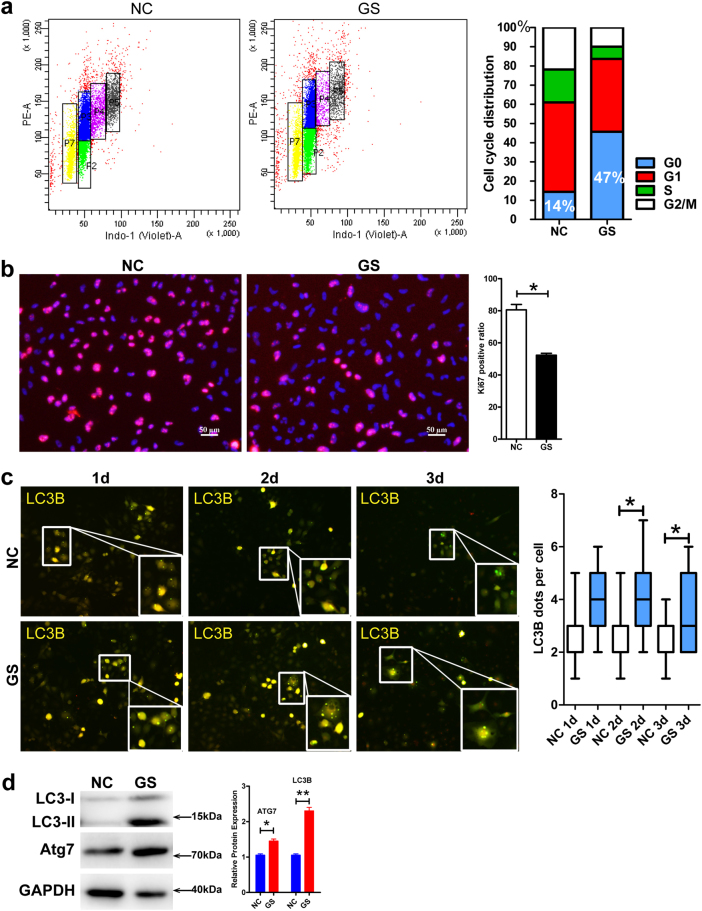
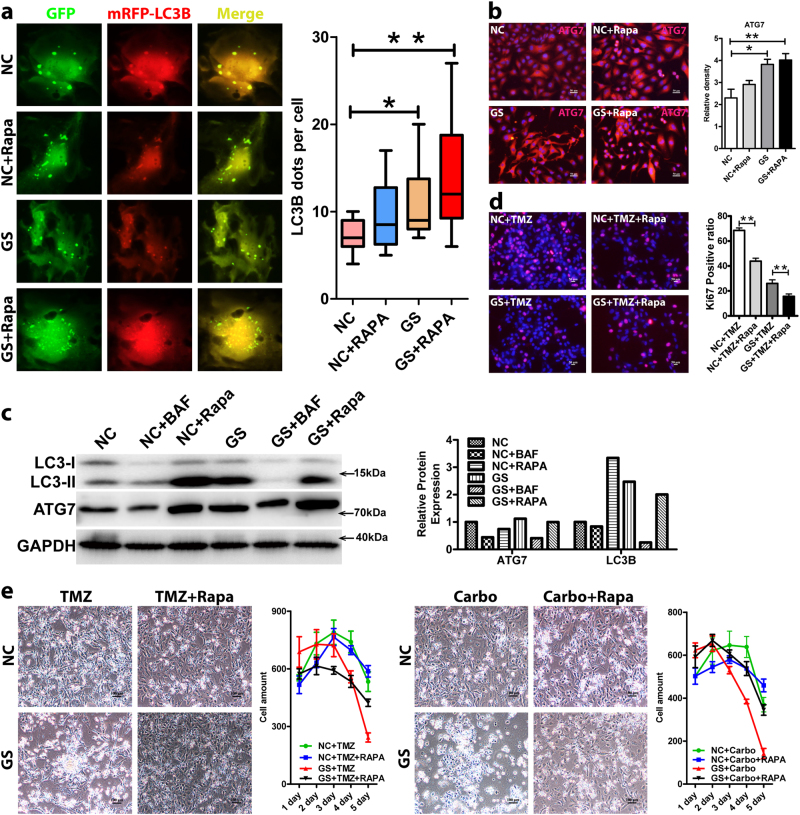
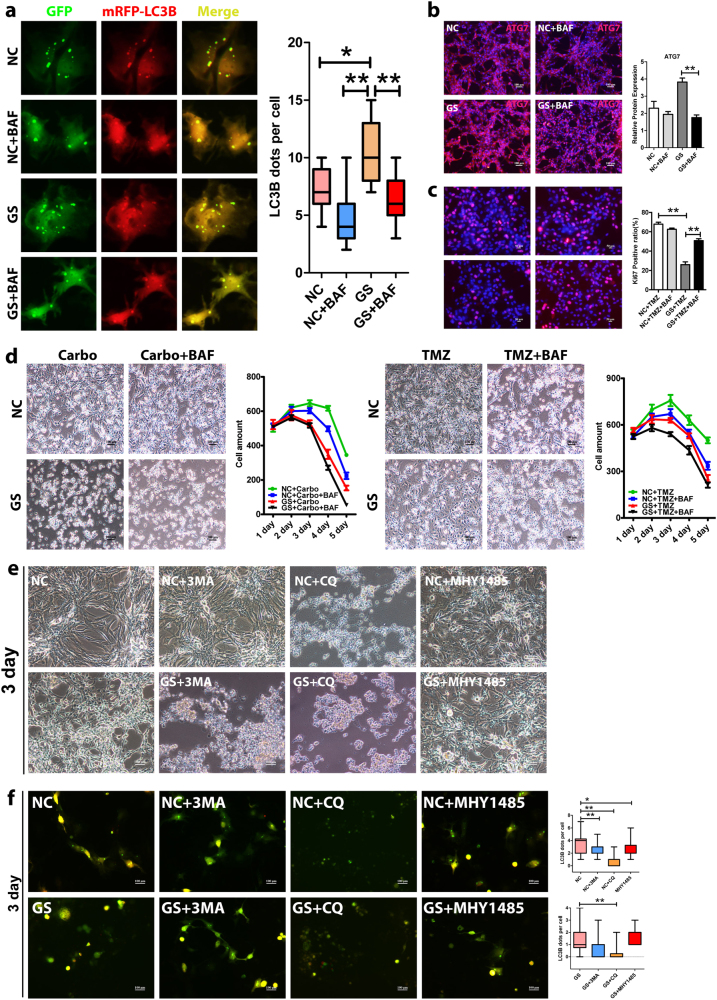
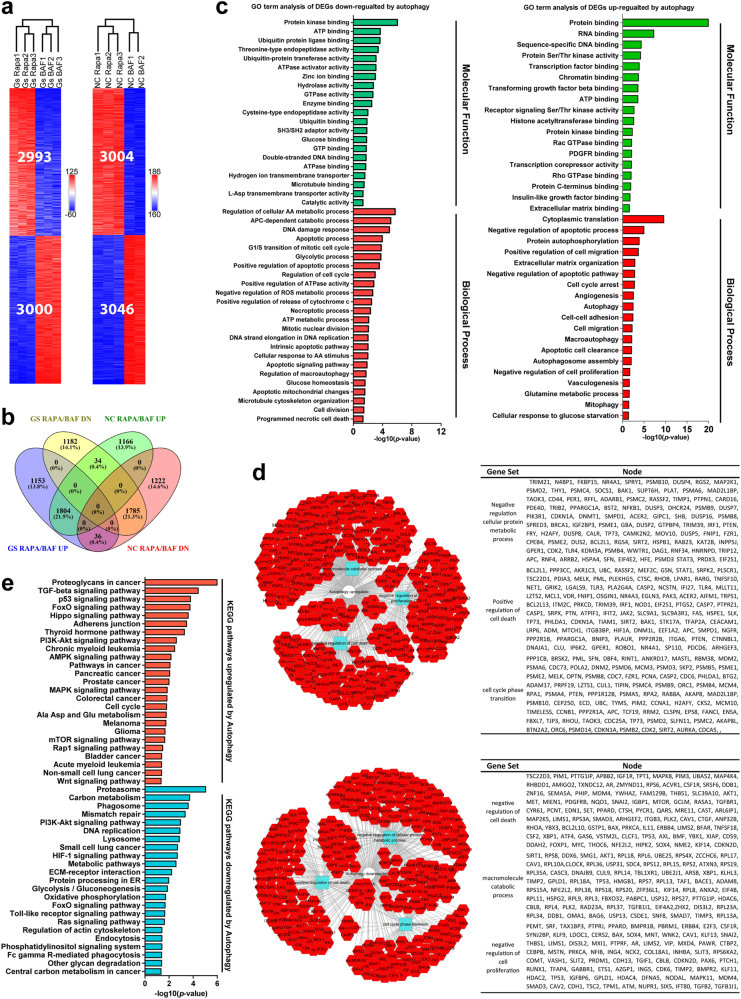
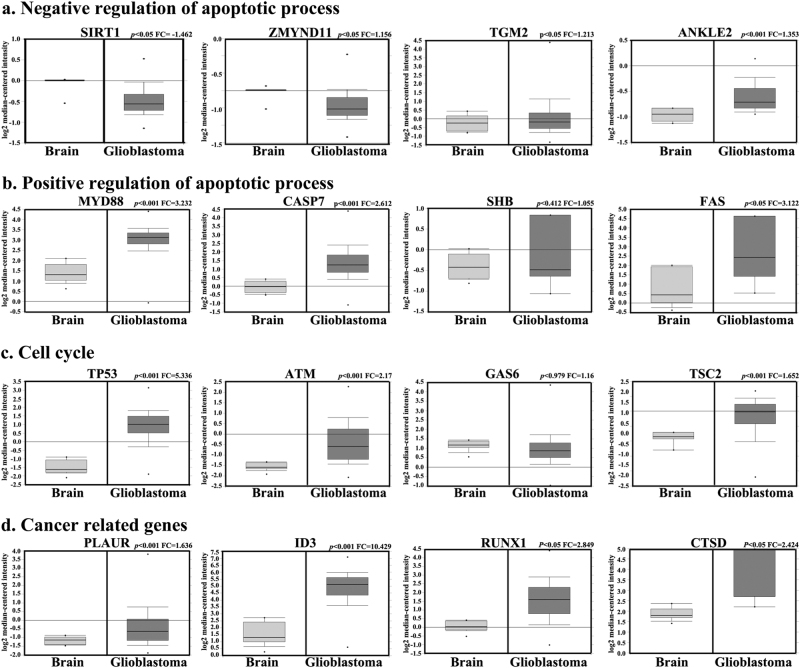
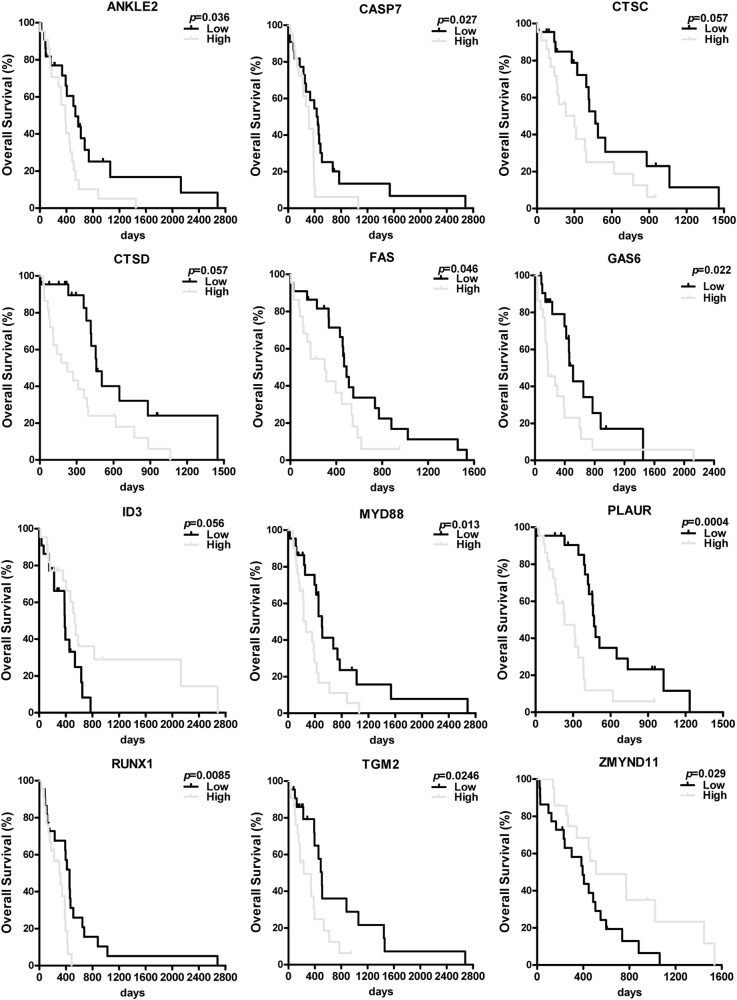
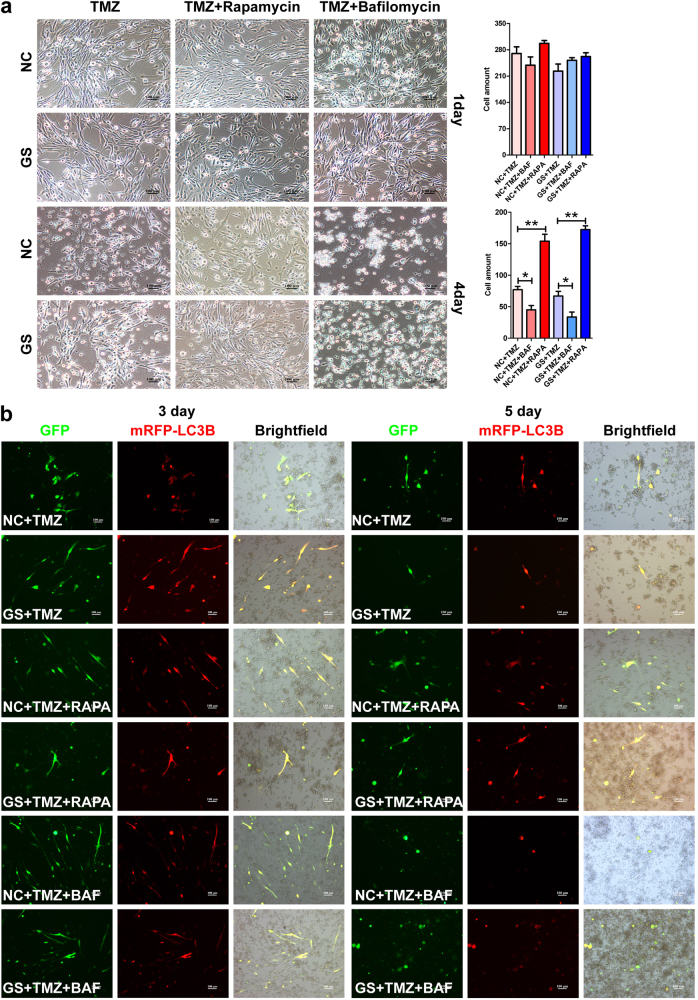
Metabolic reprogramming is pivotal to sustain cancer growth and progression. As such dietary restriction therapy represents a promising approach to starve and treat cancers. Nonetheless, tumors are dynamic and heterogeneous populations of cells with metabolic activities modulated by spatial and temporal contexts. Autophagy is a major pathway controlling cell metabolism. It can downregulate cell metabolism, leading to cancer cell quiescence, survival, and chemoresistance. To understand treatment dynamics and provide rationales for better future therapeutic strategies, we investigated whether and how autophagy is involved in the chemo-cytotoxicity and -resistance using two commonly used human glioblastoma (GBM) cell lines U87 and U251 together with primary cancer cells from the GBM patients. Our results suggest that autophagy mediates chemoresistance through reprogramming cancer cell metabolism and promoting quiescence and survival. Further unbiased transcriptome profiling identified a number of clinically relevant pathways and genes, strongly correlated with TCGA data. Our analyses have not only reported many well-known tumor players, but also uncovered a number of genes that were not previously implicated in cancers and/or GBM. The known functions of these genes are highly suggestive. It would be of high interest to investigate their potential involvement in GBM tumorigenesis, progression, and/or drug resistance. Taken together, our results suggest that autophagy inhibition could be a viable approach to aid GBM chemotherapy and combat drug resistance.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
