

Advances in spinal muscular atrophy therapeutics
- PMID: 29434670
- PMCID: PMC5802612
- DOI: 10.1177/1756285618754501
Advances in spinal muscular atrophy therapeutics
Abstract
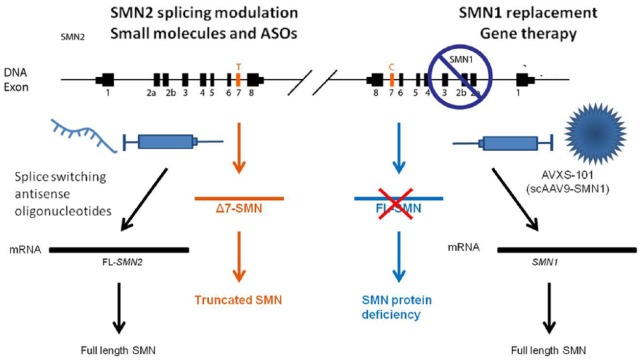
Spinal muscular atrophy (SMA) is a progressive, recessively inherited neuromuscular disease, characterized by the degeneration of lower motor neurons in the spinal cord and brainstem, which leads to weakness and muscle atrophy. SMA currently represents the most common genetic cause of infant death. SMA is caused by the lack of survival motor neuron (SMN) protein due to mutations, which are often deletions, in the SMN1 gene. In the absence of treatments able to modify the disease course, a considerable burden falls on patients and their families. Greater knowledge of the molecular basis of SMA pathogenesis has fuelled the development of potential therapeutic approaches, which are illustrated here. Nusinersen, a modified antisense oligonucleotide that modulates the splicing of the SMN2 mRNA transcript, is the first approved drug for all types of SMA. Moreover, the first gene therapy clinical trial using adeno-associated virus (AAV) vectors encoding SMN reported positive results in survival and motor milestones achievement. In addition, other strategies are in the pipeline, including modulation of SMN2 transcripts, neuroprotection, and targeting an increasing number of other peripheral targets, including the skeletal muscle. Based on this premise, it is reasonable to expect that therapeutic approaches aimed at treating SMA will soon be changed, and improved, in a meaningful way. We discuss the challenges with regard to the development of novel treatments for patients with SMA, and depict the current and future scenarios as the field enters into a new era of promising effective treatments.
Keywords: antisense oligonucleotides; gene therapy; neuromuscular disease; spinal muscular atrophy.
Conflict of interest statement
Conflict of interest statement: The authors declare that there is no conflict of interest.
Figures
References
-
- Oskoui M, Darras BT, De Vivo DC. Spinal muscular atrophy: 125 years later and on the verge of a cure. In: Sumner CJ, Paushkin S, Ko CPN. (eds) Spinal muscular atrophy. Disease mechanisms and therapy. 1st ed. London, UK: Academic Press, 2017, pp.3–17.
-
- Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995; 80: 155–165. - PubMed
-
- Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 1999; 8: 1177–1183. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
