

Excitonic splittings in molecular dimers: why static ab initio calculations cannot match them
- PMID: 29435210
- PMCID: PMC5802277
- DOI: 10.1039/c5sc02546j
Excitonic splittings in molecular dimers: why static ab initio calculations cannot match them
Abstract
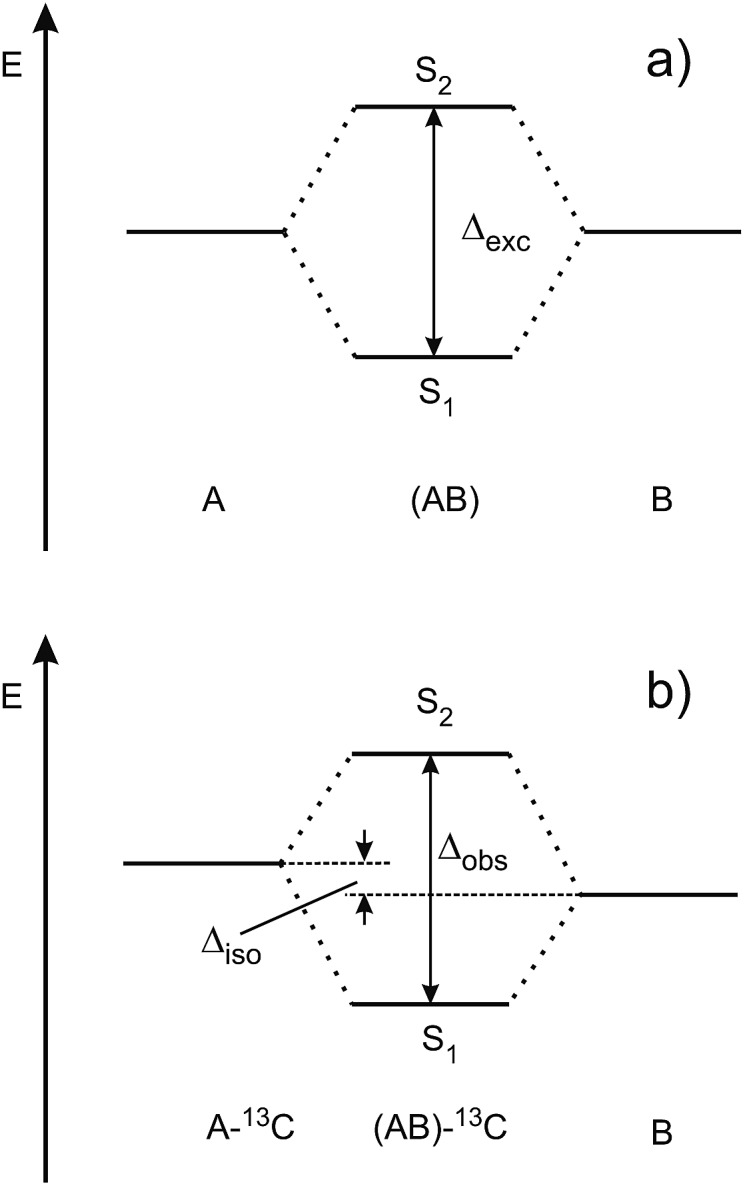
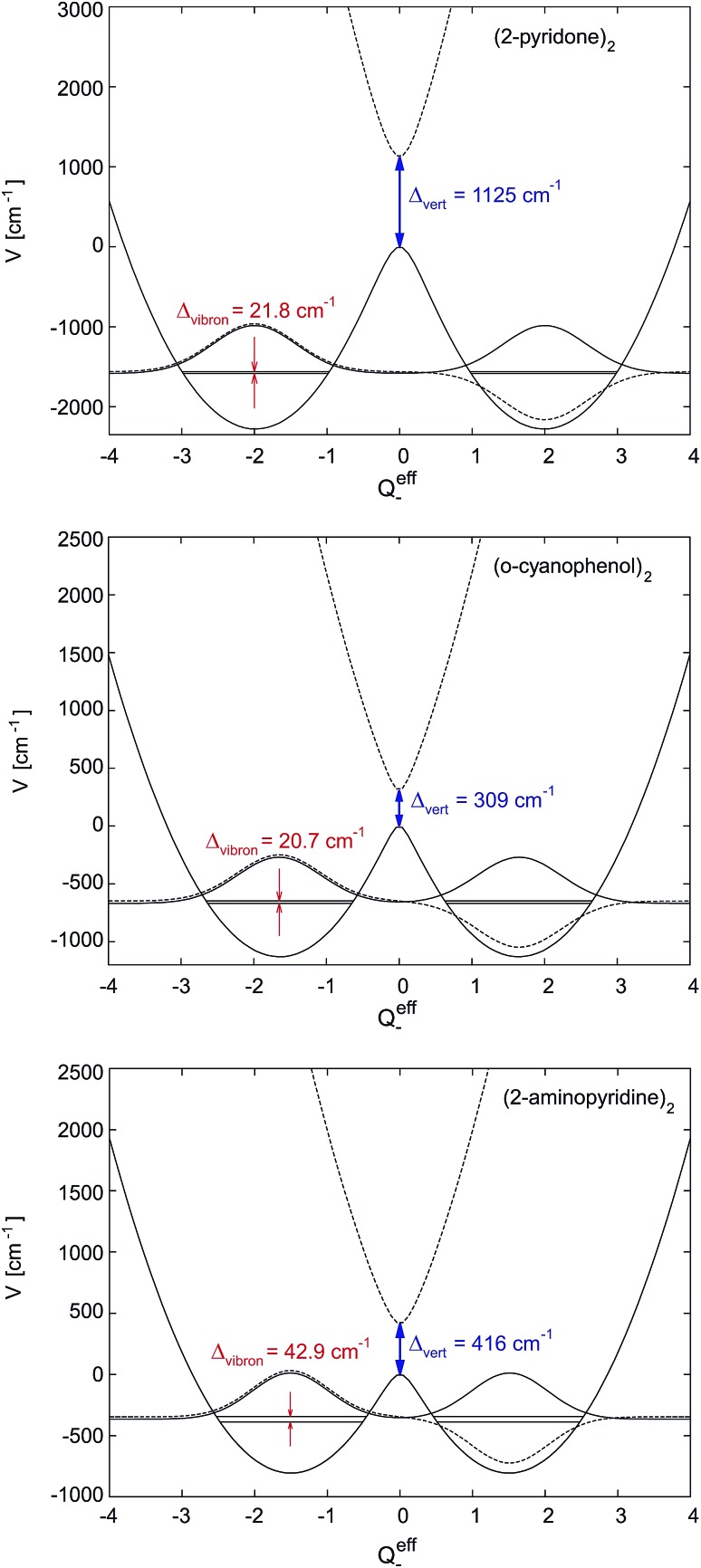
After decades of research on molecular excitons, only few molecular dimers are available on which exciton and vibronic coupling theories can be rigorously tested. In centrosymmetric H-bonded dimers consisting of identical (hetero)aromatic chromophores, the monomer electronic transition dipole moment vectors subtract or add, yielding S0 → S1 and S0 → S2 transitions that are symmetry-forbidden or -allowed, respectively. Symmetry breaking by 12C/13C or H/D isotopic substitution renders the forbidden transition weakly allowed. The excitonic coupling (Davydov splitting) can then be measured between the S0 → S1 and S0 → S2 vibrationless bands. We discuss the mass-specific excitonic spectra of five H-bonded dimers that are supersonically cooled to a few K and investigated using two-color resonant two-photon ionization spectroscopy. The excitonic splittings Δcalc predicted by ab initio methods are 5-25 times larger than the experimental excitonic splittings Δexp. The purely electronic ab initio splittings need to be reduced ("quenched"), reflecting the coupling of the electronic transition to the optically active vibrations of the monomers. The so-called quenching factors Γ < 1 can be determined from experiment (Γexp) and/or calculation (Γcalc). The vibronically quenched splittings Γ·Δcalc are found to nicely reproduce the experimental exciton splittings.
Figures




References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources