

Interpreting Genetic Variants in Titin in Patients With Muscle Disorders
- PMID: 29435569
- PMCID: PMC5885217
- DOI: 10.1001/jamaneurol.2017.4899
Interpreting Genetic Variants in Titin in Patients With Muscle Disorders
Erratum in
-
Incorrect Degree.JAMA Neurol. 2018 Nov 1;75(11):1443. doi: 10.1001/jamaneurol.2018.2391. JAMA Neurol. 2018. PMID: 30083759 Free PMC article. No abstract available.
Abstract
Importance: Mutations in the titin gene (TTN) cause a wide spectrum of genetic diseases. The interpretation of the numerous rare variants identified in TTN is a difficult challenge given its large size.
Objective: To identify genetic variants in titin in a cohort of patients with muscle disorders.
Design, setting, and participants: In this case series, 9 patients with titinopathy and 4 other patients with possibly disease-causing variants in TTN were identified. Titin mutations were detected through targeted resequencing performed on DNA from 504 patients with muscular dystrophy, congenital myopathy, or other skeletal muscle disorders. Patients were enrolled from 10 clinical centers in April 2012 to December 2013. All of them had not received a diagnosis after undergoing an extensive investigation, including Sanger sequencing of candidate genes. The data analysis was performed between September 2013 and January 2017. Sequencing data were analyzed using an internal custom bioinformatics pipeline.
Main outcomes and measures: The identification of novel mutations in the TTN gene and novel patients with titinopathy. We performed an evaluation of putative causative variants in the TTN gene, combining genetic, clinical, and imaging data with messenger RNA and/or protein studies.
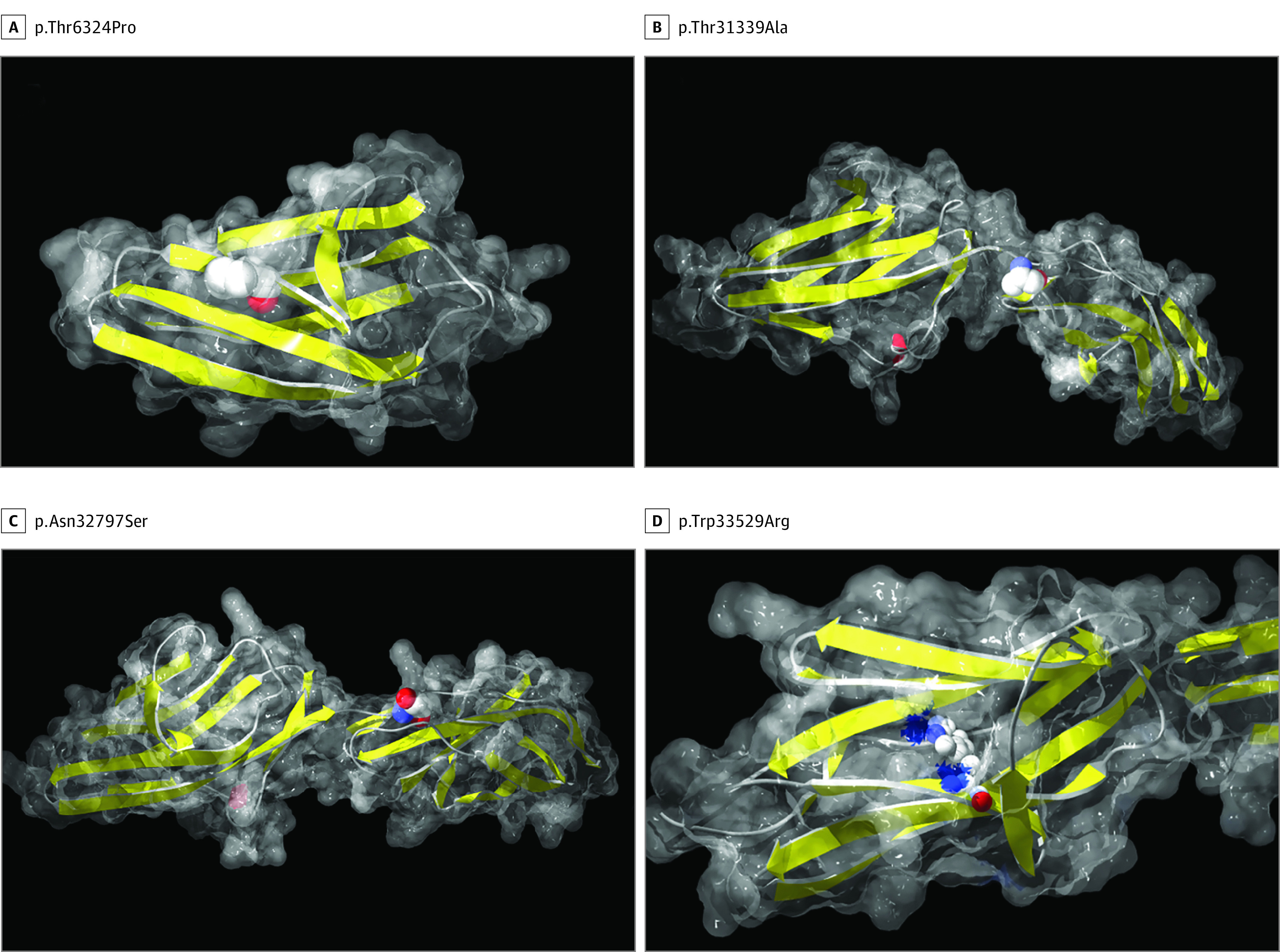
Results: Of the 9 novel patients with titinopathy, 5 (55.5%) were men and the mean (SD) age at onset was 25 (15.8) years (range, 0-46 years). Of the 4 other patients (3 men and 1 woman) with possibly disease-causing TTN variants, 2 (50%) had a congenital myopathy and 2 (50%) had a slowly progressive distal myopathy with onset in the second decade. Most of the identified mutations were previously unreported. However, all the variants, even the already described mutations, require careful clinical and molecular evaluation of probands and relatives. Heterozygous truncating variants or unique missense changes are not sufficient to make a diagnosis of titinopathy.
Conclusions and relevance: The interpretation of TTN variants often requires further analyses, including a comprehensive evaluation of the clinical phenotype (deep phenotyping) as well as messenger RNA and protein studies. We propose a specific workflow for the clinical interpretation of genetic findings in titin.
Conflict of interest statement
Figures


Comment in
-
Understanding Titin Variants in the Age of Next-Generation Sequencing-A Titanic Challenge.JAMA Neurol. 2018 May 1;75(5):539-540. doi: 10.1001/jamaneurol.2017.3068. JAMA Neurol. 2018. PMID: 29435554 No abstract available.
References
-
- Bang ML, Centner T, Fornoff F, et al. . The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res. 2001;89(11):1065-1072. - PubMed
-
- Gerull B. The rapidly evolving role of titin in cardiac physiology and cardiomyopathy. Can J Cardiol. 2015;31(11):1351-1359. - PubMed
-
- Chauveau C, Rowell J, Ferreiro A. A rising titan: TTN review and mutation update. Hum Mutat. 2014;35(9):1046-1059. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
