

Breast cancer family history and allele-specific DNA methylation in the legacy girls study
- PMID: 29436922
- PMCID: PMC5997170
- DOI: 10.1080/15592294.2018.1435243
Breast cancer family history and allele-specific DNA methylation in the legacy girls study
Abstract
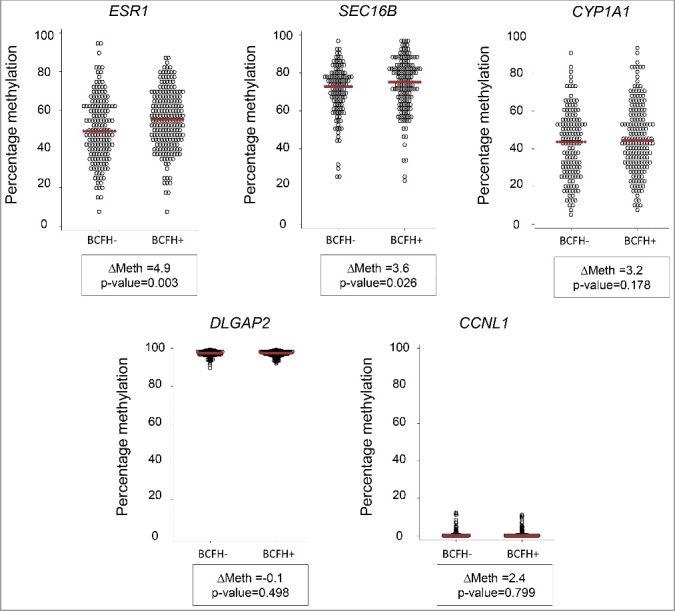
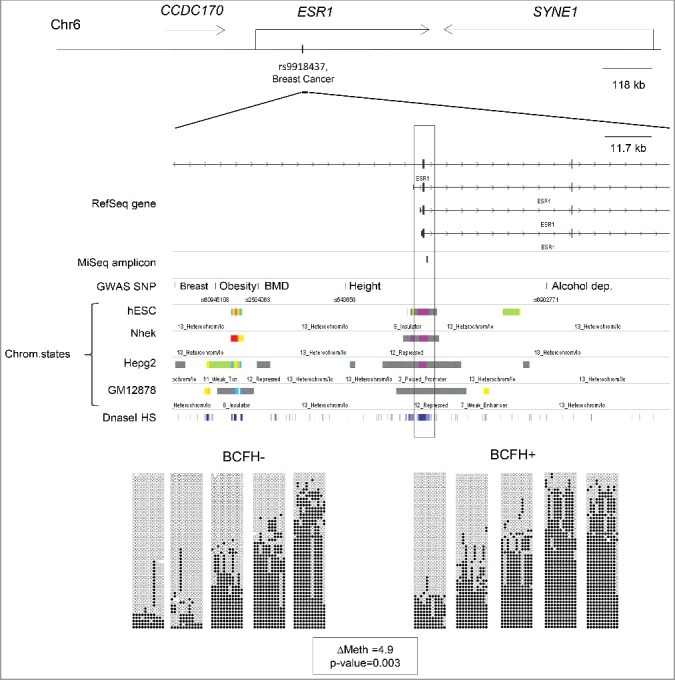
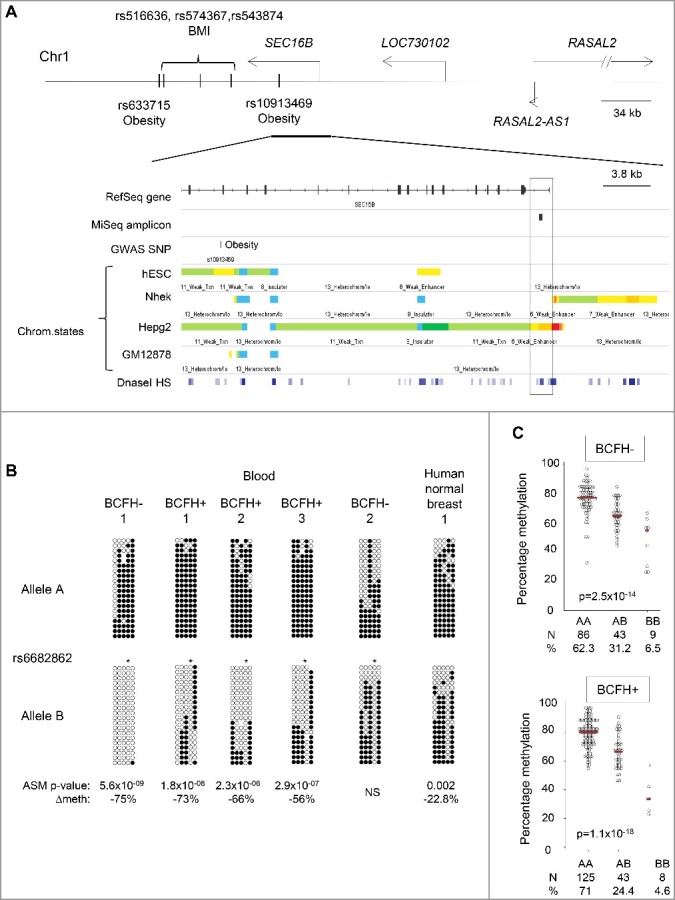
Family history, a well-established risk factor for breast cancer, can have both genetic and environmental contributions. Shared environment in families as well as epigenetic changes that also may be influenced by shared genetics and environment may also explain familial clustering of cancers. Epigenetic regulation, such as DNA methylation, can change the activity of a DNA segment without a change in the sequence; environmental exposures experienced across the life course can induce such changes. However, genetic-epigenetic interactions, detected as methylation quantitative trait loci (mQTLs; a.k.a. meQTLs) and haplotype-dependent allele-specific methylation (hap-ASM), can also contribute to inter-individual differences in DNA methylation patterns. To identify differentially methylated regions (DMRs) associated with breast cancer susceptibility, we examined differences in white blood cell DNA methylation in 29 candidate genes in 426 girls (ages 6-13 years) from the LEGACY Girls Study, 239 with and 187 without a breast cancer family history (BCFH). We measured methylation by targeted massively parallel bisulfite sequencing (bis-seq) and observed BCFH DMRs in two genes: ESR1 (Δ4.9%, P = 0.003) and SEC16B (Δ3.6%, P = 0.026), each of which has been previously implicated in breast cancer susceptibility and pubertal development. These DMRs showed high inter-individual variability in methylation, suggesting the presence of mQTLs/hap-ASM. Using single nucleotide polymorphisms data in the bis-seq amplicon, we found strong hap-ASM in SEC16B (with allele specific-differences ranging from 42% to 74%). These findings suggest that differential methylation in genes relevant to breast cancer susceptibility may be present early in life, and that inherited genetic factors underlie some of these epigenetic differences.
Keywords: Breast cancer family history; DNA methylation; mQTL; white blood cells, childhood and adolescent cohort.
Figures
References
-
- Cancer CGoHFiB Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58 209 women with breast cancer and 101 986 women without the disease. The Lancet. 2001;358:1389–1399. doi:http://dx.doi.org/10.1016/S0140-6736(01)06524-2. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous