

hsa-mir183/EGR1-mediated regulation of E2F1 is required for CML stem/progenitor cell survival
- PMID: 29437554
- PMCID: PMC6027092
- DOI: 10.1182/blood-2017-05-783845
hsa-mir183/EGR1-mediated regulation of E2F1 is required for CML stem/progenitor cell survival
Abstract
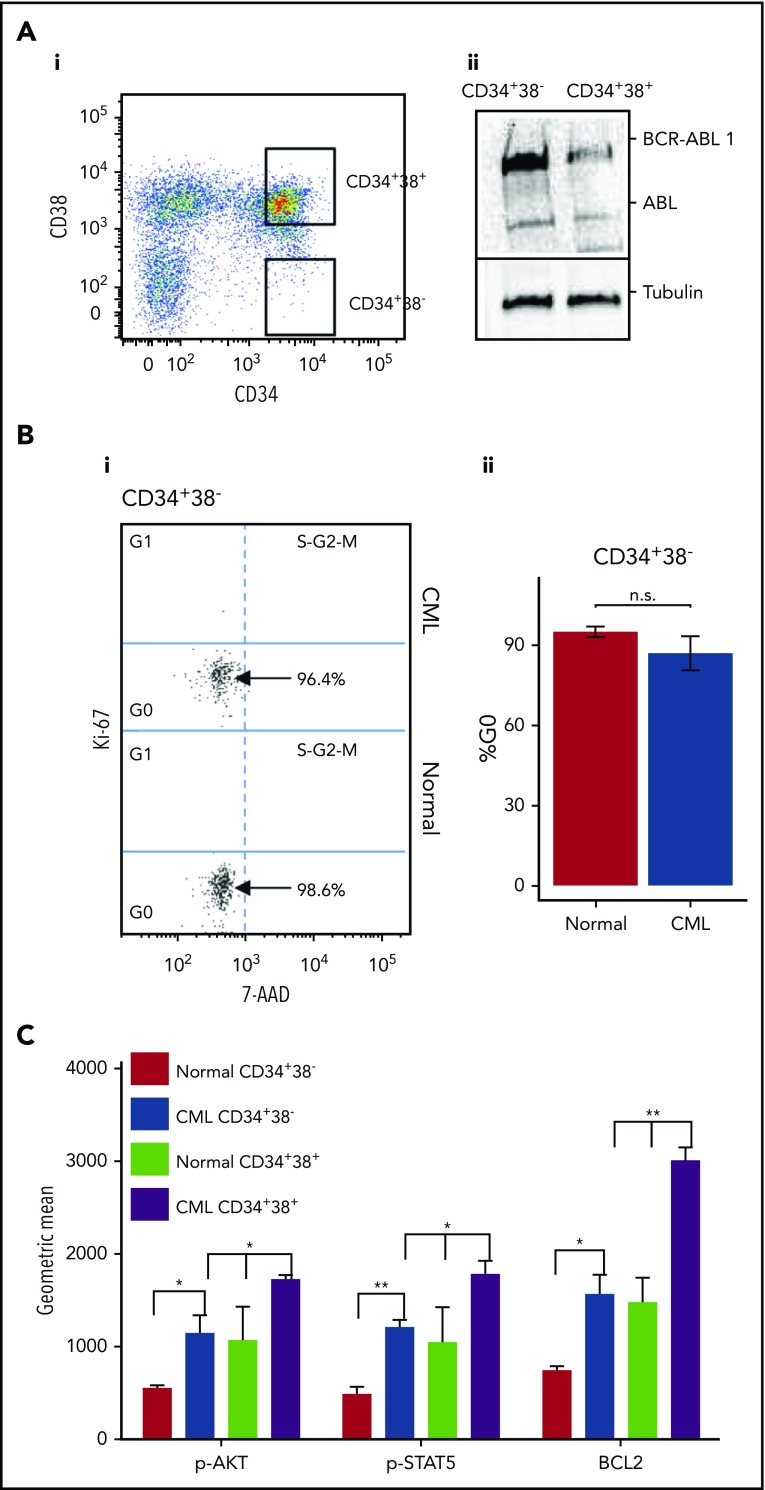
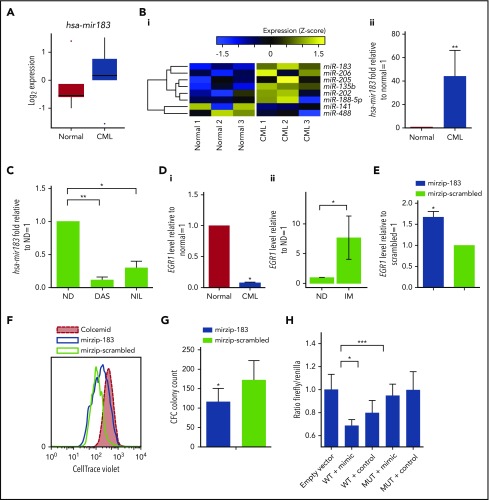
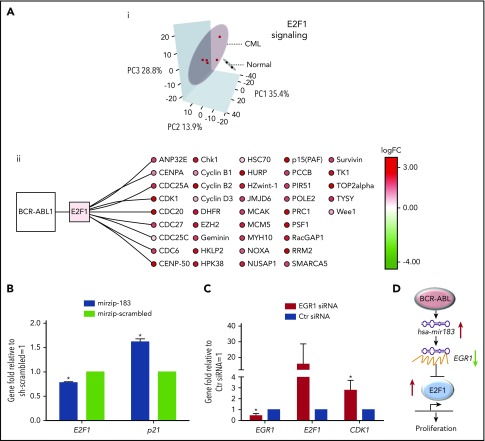
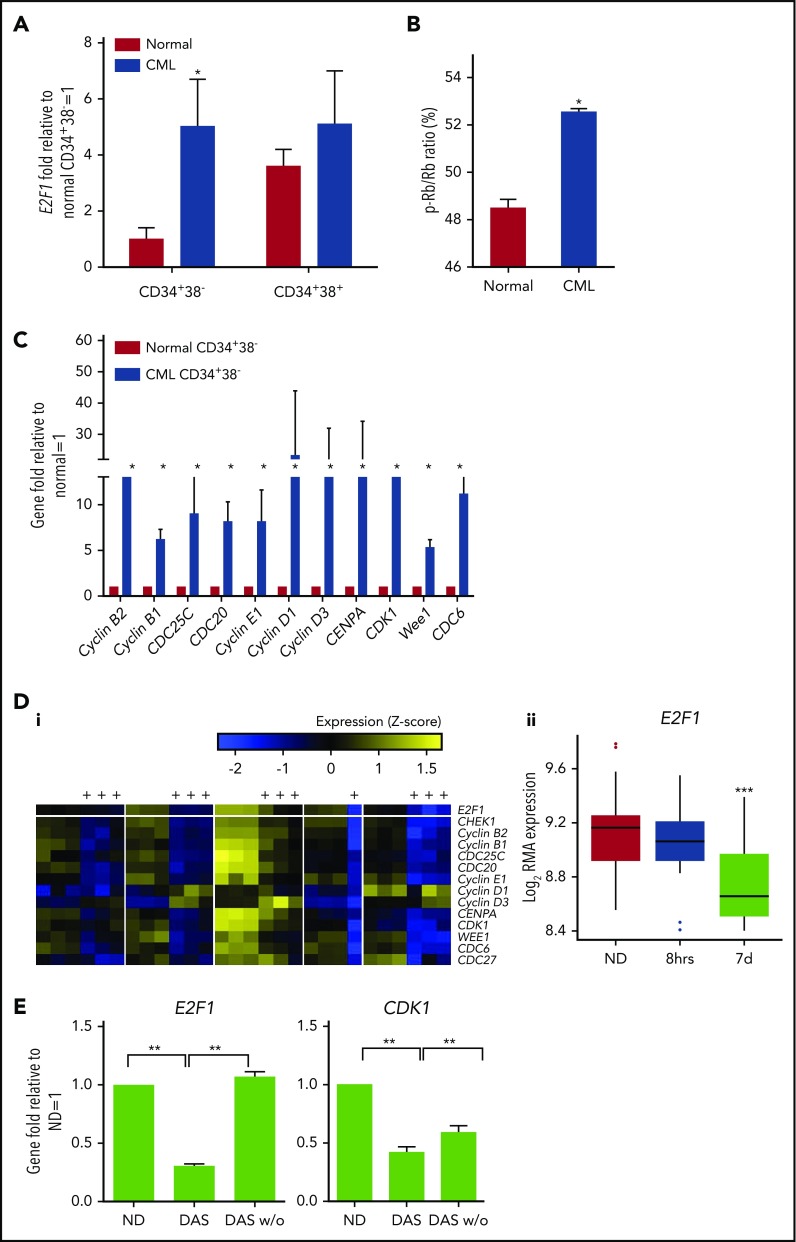
Chronic myeloid leukemia (CML) stem/progenitor cells (SPCs) express a transcriptional program characteristic of proliferation, yet can achieve and maintain quiescence. Understanding the mechanisms by which leukemic SPCs maintain quiescence will help to clarify how they persist during long-term targeted treatment. We have identified a novel BCR-ABL1 protein kinase-dependent pathway mediated by the upregulation of hsa-mir183, the downregulation of its direct target early growth response 1 (EGR1), and, as a consequence, upregulation of E2F1. We show here that inhibition of hsa-mir183 reduced proliferation and impaired colony formation of CML SPCs. Downstream of this, inhibition of E2F1 also reduced proliferation of CML SPCs, leading to p53-mediated apoptosis. In addition, we demonstrate that E2F1 plays a pivotal role in regulating CML SPC proliferation status. Thus, for the first time, we highlight the mechanism of hsa-mir183/EGR1-mediated E2F1 regulation and demonstrate this axis as a novel, critical factor for CML SPC survival, offering new insights into leukemic stem cell eradication.
© 2018 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
Comment in
-
Et tu, E2F1? The assassins of CML stem cells.Blood. 2018 Apr 5;131(14):1499-1500. doi: 10.1182/blood-2018-02-833699. Blood. 2018. PMID: 29622533 No abstract available.
References
-
- Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2(5):561-566. - PubMed
-
- Bhatia R, Holtz M, Niu N, et al. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood. 2003;101(12):4701-4707. - PubMed
-
- Mahon FX, Réa D, Guilhot J, et al. ; Intergroupe Français des Leucémies Myéloïdes Chroniques. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029-1035. - PubMed
-
- Graham SM, Jørgensen HG, Allan E, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319-325. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
