

HO-1hi patrolling monocytes protect against vaso-occlusion in sickle cell disease
- PMID: 29437594
- PMCID: PMC5887768
- DOI: 10.1182/blood-2017-12-819870
HO-1hi patrolling monocytes protect against vaso-occlusion in sickle cell disease
Abstract
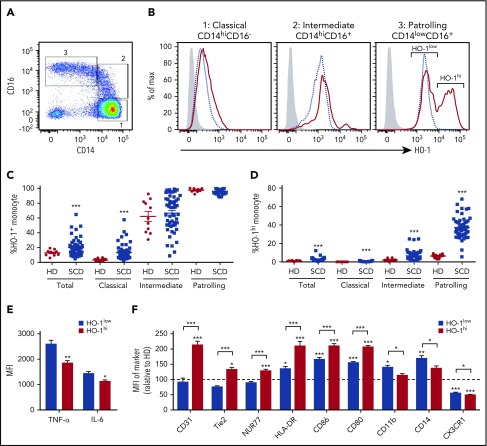
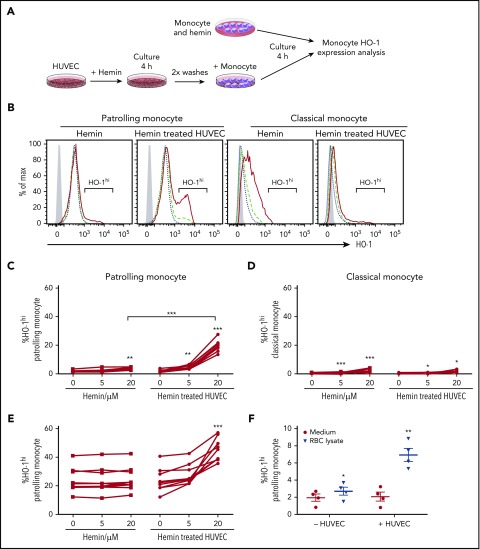
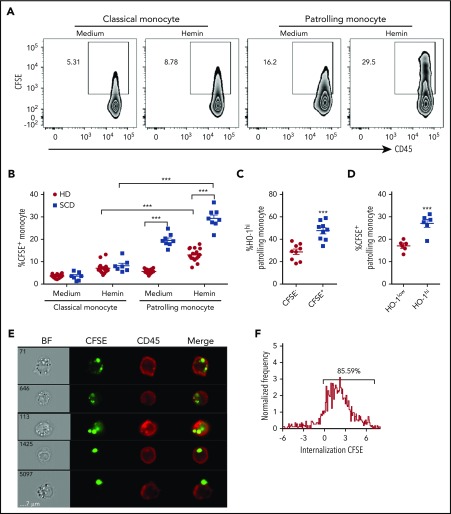
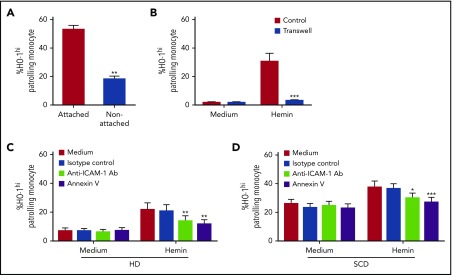
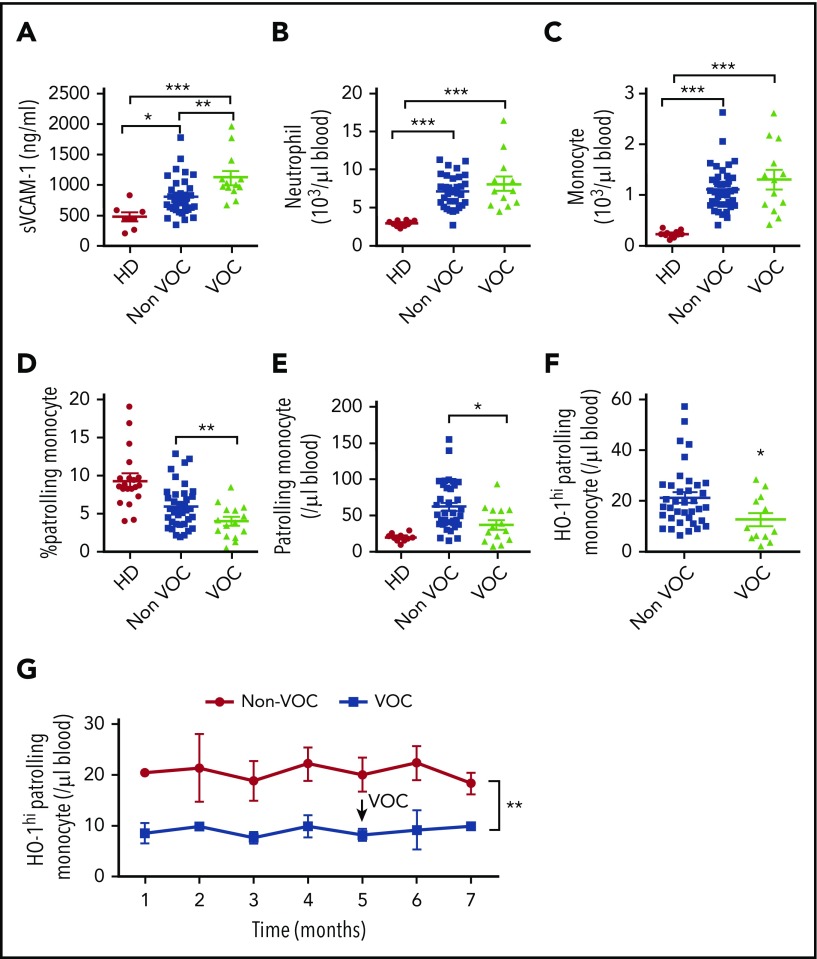
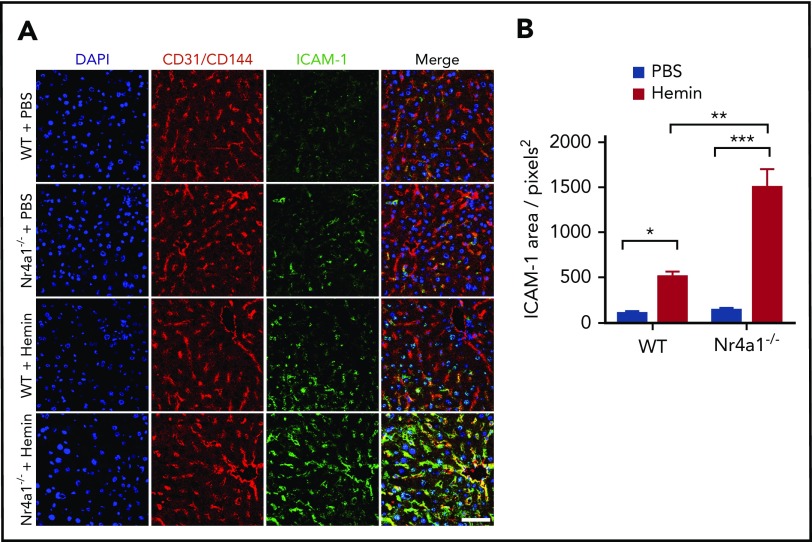
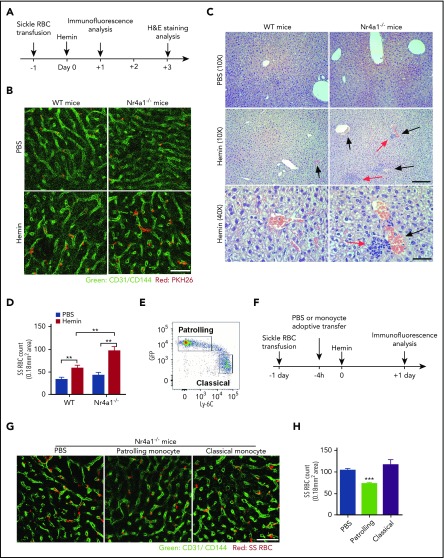
Patients with sickle cell disease (SCD) suffer from intravascular hemolysis associated with vascular injury and dysfunction in mouse models, and painful vaso-occlusive crisis (VOC) involving increased attachment of sickle erythrocytes and activated leukocytes to damaged vascular endothelium. Patrolling monocytes, which normally scavenge damaged cells and debris from the vasculature, express higher levels of anti-inflammatory heme oxygenase 1 (HO-1), a heme degrading enzyme. Here, we show that HO-1-expressing patrolling monocytes protect SCD vasculature from ongoing hemolytic insult and vaso-occlusion. We found that a mean 37% of patrolling monocytes from SCD patients express very high levels of HO-1 (HO-1hi) vs 6% in healthy controls and demonstrated that HO-1hi expression was dependent on uptake of heme-exposed endothelium. SCD patients with a recent VOC episode had lower numbers of HO-1hi patrolling monocytes. Heme-mediated vaso-occlusion by mouse SCD red blood cells was exacerbated in mice lacking patrolling monocytes, and reversed following transfer of patrolling monocytes. Altogether, these data indicate that SCD patrolling monocytes remove hemolysis-damaged endothelial cells, resulting in HO-1 upregulation and dampening of VOC, and that perturbation in patrolling monocyte numbers resulting in lower numbers of HO-1hi patrolling monocyte may predispose SCD patients to VOC. These data suggest that HO-1hi patrolling monocytes are key players in VOC pathophysiology and have potential as therapeutic targets for VOC.
© 2018 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
Comment in
-
Patrolling the endothelium in sickle cell disease.Blood. 2018 Apr 5;131(14):1503-1505. doi: 10.1182/blood-2018-02-832139. Blood. 2018. PMID: 29622536 No abstract available.
References
-
- Gladwin MT. Cardiovascular complications and risk of death in sickle-cell disease. Lancet. 2016;387(10037):2565-2574. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
