

Argininemia as a cause of severe chronic stunting and partial growth hormone deficiency (PGHD): A case report
- PMID: 29443755
- PMCID: PMC5839826
- DOI: 10.1097/MD.0000000000009880
Argininemia as a cause of severe chronic stunting and partial growth hormone deficiency (PGHD): A case report
Abstract
Rationale: Argininemia is an autosomal recessive inherited disorder of the urea cycle. Because of its atypical symptoms in early age, diagnosis can be delayed until the typical chronic manifestations - including spastic diplegia, deterioration in cognitive function, and epilepsy - appear in later childhood.
Patient concerns: A Chinese boy initially presented with severe stunting and partial growth hormone deficiency (PGHD) at 3 years old and was initially treated with growth hormone replacement therapy. Seven years later (at 10 years old), he presented with spastic diplegia, cognitive function lesions, epilepsy, and peripheral neuropathy.
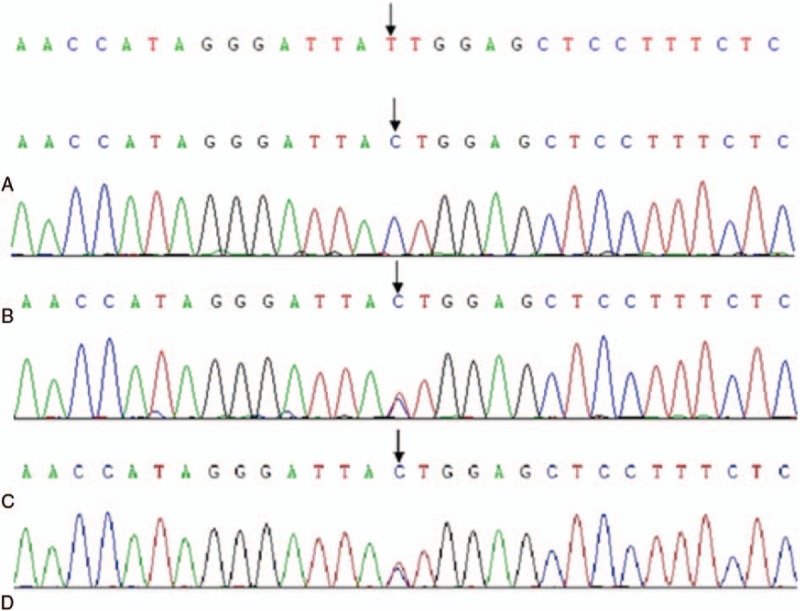
Diagnoses: Ultimately, the patient was diagnosed with argininemia with homozygous mutation (c.32T>C) of the ARG1 gene at 10 years old. Blood tests showed mildly elevated blood ammonia and creatine kinase, and persistently elevated bilirubin.
Interventions: Protein intake was limited to 0.8 g/kg/day, citrulline (150-200 mg [kg d]) was prescribed.
Outcomes: The patient's mental state and vomiting had improved after 3 months treatment. At 10 years and 9 month old, his height and weight had reached 121cm and 22kg, respectively, but his spastic diplegia symptoms had not improved.
Lessons: This case demonstrates that stunting and PGHD that does not respond to growth hormone replacement therapy might hint at inborn errors of metabolism (IEM). IEM should also be considered in patients with persistently elevated bilirubin with or without abnormal liver transaminase, as well as elevated blood ammonia and creatine kinase, in the absence of hepatic disease.
Conflict of interest statement
The authors have no conflicts of interest to disclose.
Figures

References
-
- Nagata N, Matsuda I, Oyanagi K. Estimated frequency of urea cycle enzymopathies in Japan. Am J Med Genet 2010;39:228–9. - PubMed
-
- Testai FD, Gorelick PB. Inherited metabolic disorders and stroke part 2: homocystinuria, organic acidurias, and urea cycle disorders. Arch Neurol 2010;67:148–53. - PubMed
-
- Crombez EA, Cederbaum SD. Hyperargininemia due to liver arginase deficiency. Mol Genet Metab 2005;84:243–51. - PubMed
-
- Lee BH, Jin HY, Kim GH, et al. Argininemia presenting with progressive spastic diplegia. Pediatr Neurol 2011;44:218–20. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
