

Peculiar combinations of individually non-pathogenic missense mitochondrial DNA variants cause low penetrance Leber's hereditary optic neuropathy
- PMID: 29444077
- PMCID: PMC5828459
- DOI: 10.1371/journal.pgen.1007210
Peculiar combinations of individually non-pathogenic missense mitochondrial DNA variants cause low penetrance Leber's hereditary optic neuropathy
Abstract
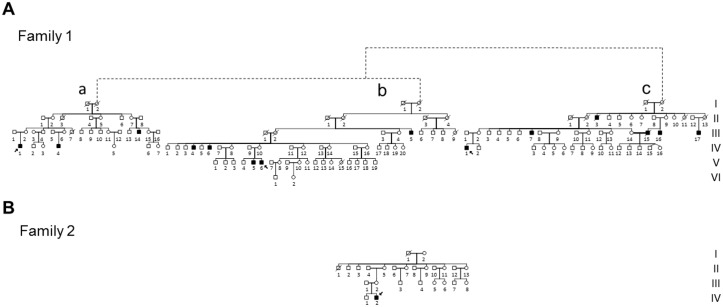
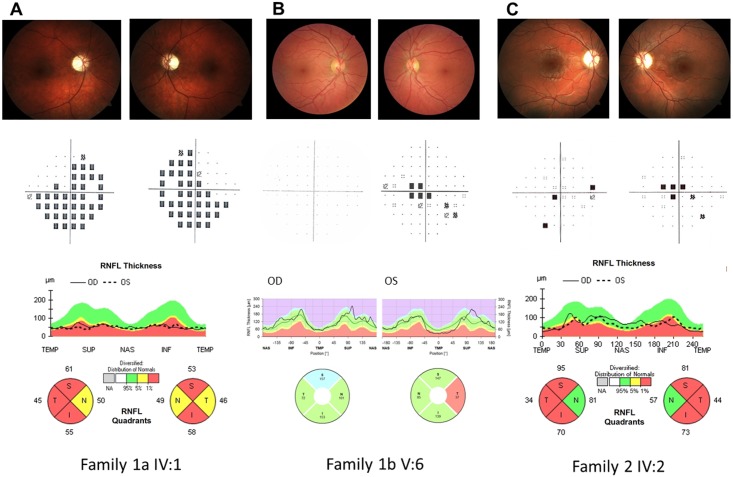
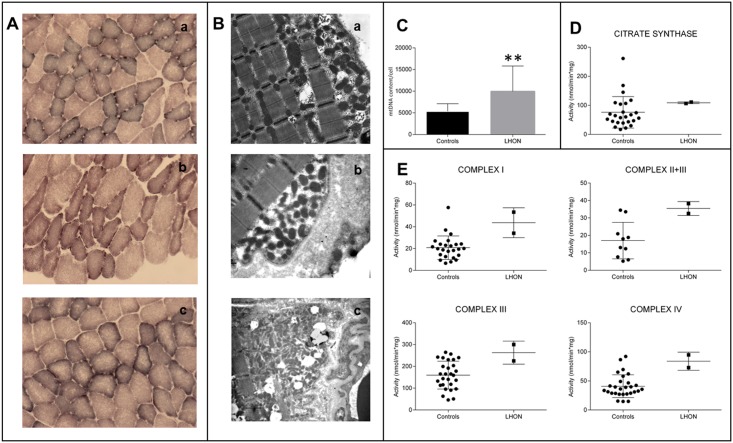
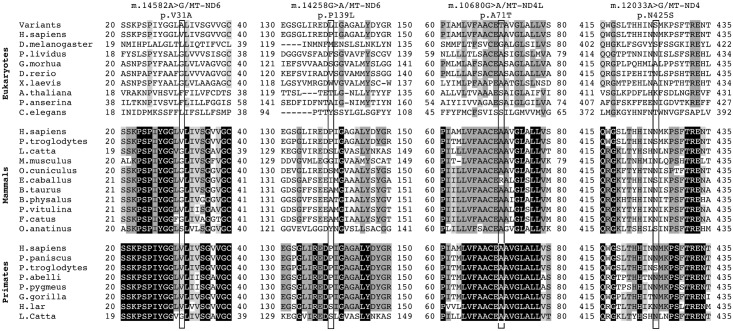
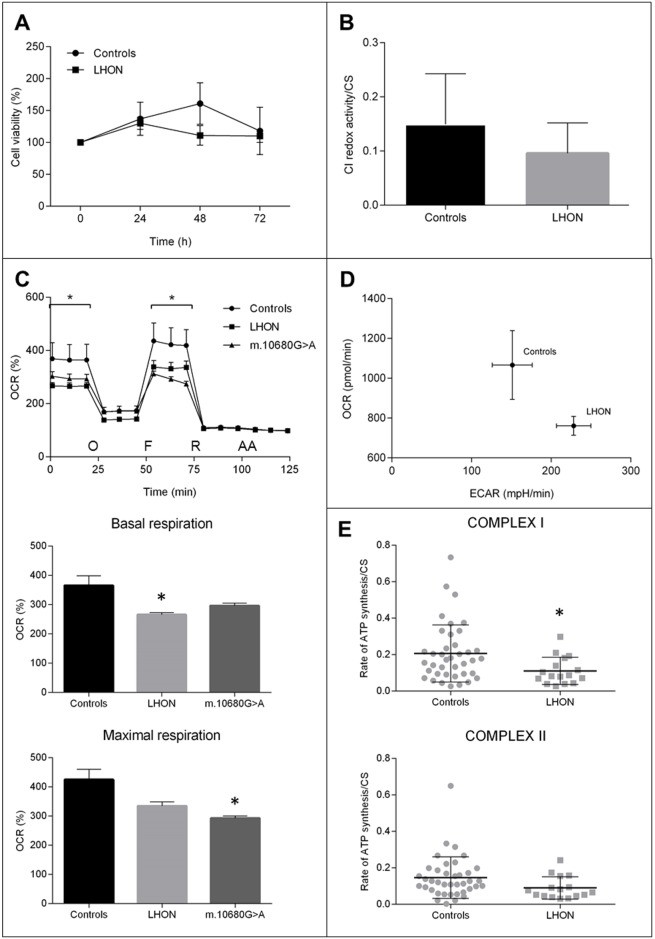
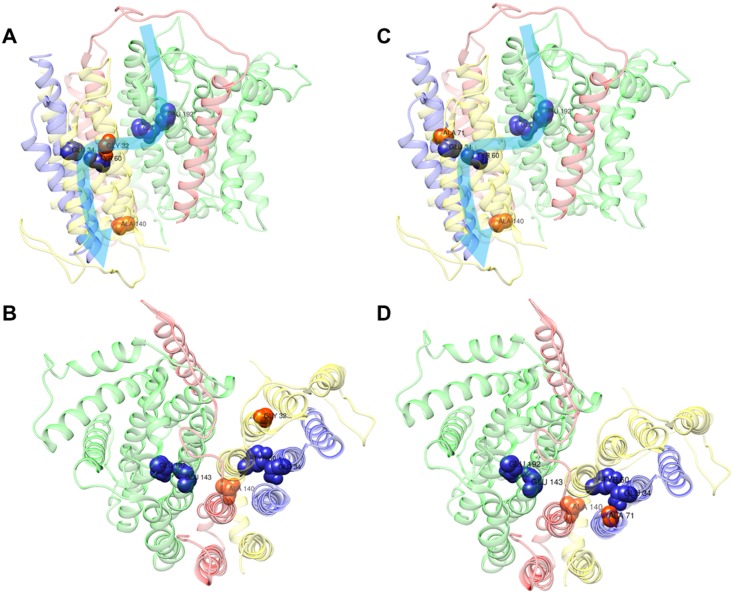
We here report on the existence of Leber's hereditary optic neuropathy (LHON) associated with peculiar combinations of individually non-pathogenic missense mitochondrial DNA (mtDNA) variants, affecting the MT-ND4, MT-ND4L and MT-ND6 subunit genes of Complex I. The pathogenic potential of these mtDNA haplotypes is supported by multiple evidences: first, the LHON phenotype is strictly inherited along the maternal line in one very large family; second, the combinations of mtDNA variants are unique to the two maternal lineages that are characterized by recurrence of LHON; third, the Complex I-dependent respiratory and oxidative phosphorylation defect is co-transferred from the proband's fibroblasts into the cybrid cell model. Finally, all but one of these missense mtDNA variants cluster along the same predicted fourth E-channel deputed to proton translocation within the transmembrane domain of Complex I, involving the ND1, ND4L and ND6 subunits. Hence, the definition of the pathogenic role of a specific mtDNA mutation becomes blurrier than ever and only an accurate evaluation of mitogenome sequence variation data from the general population, combined with functional analyses using the cybrid cell model, may lead to final validation. Our study conclusively shows that even in the absence of a clearly established LHON primary mutation, unprecedented combinations of missense mtDNA variants, individually known as polymorphisms, may lead to reduced OXPHOS efficiency sufficient to trigger LHON. In this context, we introduce a new diagnostic perspective that implies the complete sequence analysis of mitogenomes in LHON as mandatory gold standard diagnostic approach.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242: 1427–1430. - PubMed
-
- Montoya J, López-Gallardo E, Díez-Sánchez C, López-Pérez MJ, Ruiz-Pesini E. 20 years of human mtDNA pathologic point mutations: carefully reading the pathogenicity criteria. Biochim Biophys Acta. 2009;1787: 476–483. doi: 10.1016/j.bbabio.2008.09.003 - DOI - PubMed
-
- Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res. 2004;23: 53–89. doi: 10.1016/j.preteyeres.2003.10.003 - DOI - PubMed
-
- Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies—disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011;30: 81–114. doi: 10.1016/j.preteyeres.2010.11.002 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
