

Hierarchical analysis of RNA-seq reads improves the accuracy of allele-specific expression
- PMID: 29444201
- PMCID: PMC6022640
- DOI: 10.1093/bioinformatics/bty078
Hierarchical analysis of RNA-seq reads improves the accuracy of allele-specific expression
Abstract
Motivation: Allele-specific expression (ASE) refers to the differential abundance of the allelic copies of a transcript. RNA sequencing (RNA-seq) can provide quantitative estimates of ASE for genes with transcribed polymorphisms. When short-read sequences are aligned to a diploid transcriptome, read-mapping ambiguities confound our ability to directly count reads. Multi-mapping reads aligning equally well to multiple genomic locations, isoforms or alleles can comprise the majority (>85%) of reads. Discarding them can result in biases and substantial loss of information. Methods have been developed that use weighted allocation of read counts but these methods treat the different types of multi-reads equivalently. We propose a hierarchical approach to allocation of read counts that first resolves ambiguities among genes, then among isoforms, and lastly between alleles. We have implemented our model in EMASE software (Expectation-Maximization for Allele Specific Expression) to estimate total gene expression, isoform usage and ASE based on this hierarchical allocation.
Results: Methods that align RNA-seq reads to a diploid transcriptome incorporating known genetic variants improve estimates of ASE and total gene expression compared to methods that use reference genome alignments. Weighted allocation methods outperform methods that discard multi-reads. Hierarchical allocation of reads improves estimation of ASE even when data are simulated from a non-hierarchical model. Analysis of RNA-seq data from F1 hybrid mice using EMASE reveals widespread ASE associated with cis-acting polymorphisms and a small number of parent-of-origin effects.
Availability and implementation: EMASE software is available at https://github.com/churchill-lab/emase.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
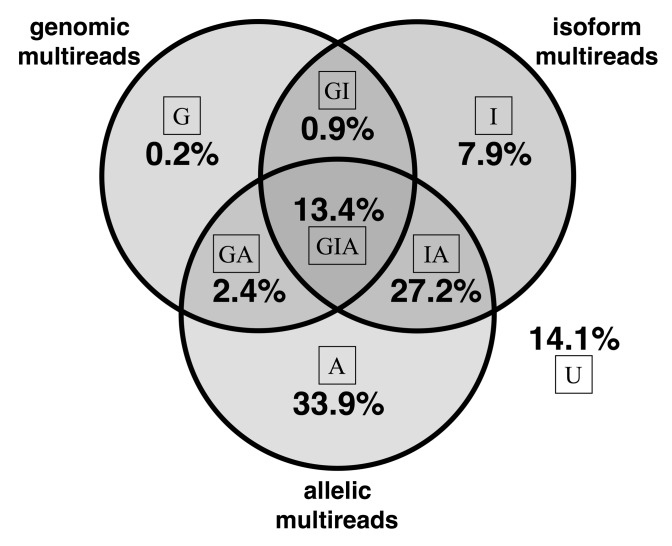
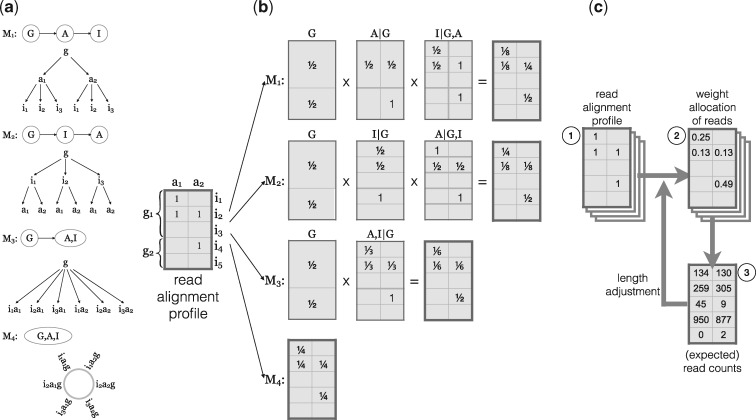
References
-
- Agresti A. (2002). Categorical Data Analysis. Wiley Series in Probability and Statistics, 2nd edn. Wiley-Interscience, New York.
-
- Bray N.L. et al. (2016) Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol., 34, 525–527. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
