

The functional genomics laboratory: functional validation of genetic variants
- PMID: 29445992
- PMCID: PMC5959958
- DOI: 10.1007/s10545-018-0146-7
The functional genomics laboratory: functional validation of genetic variants
Abstract
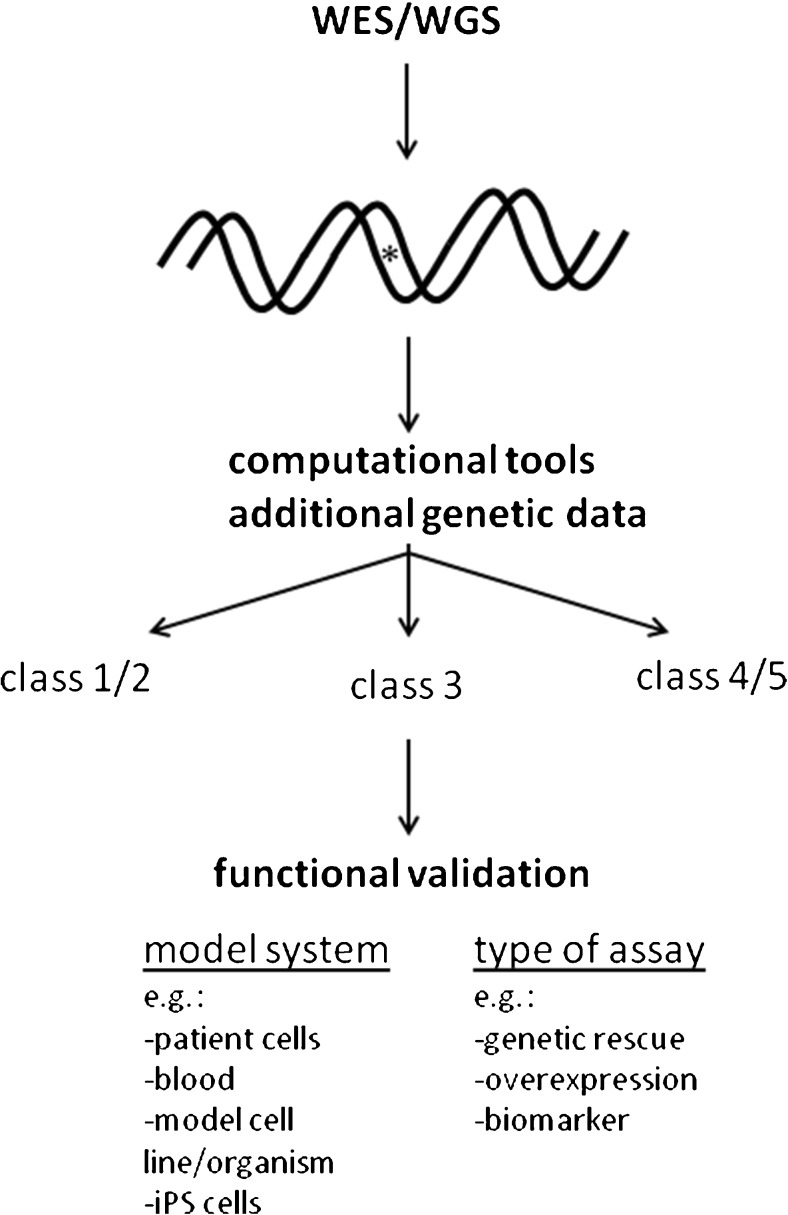
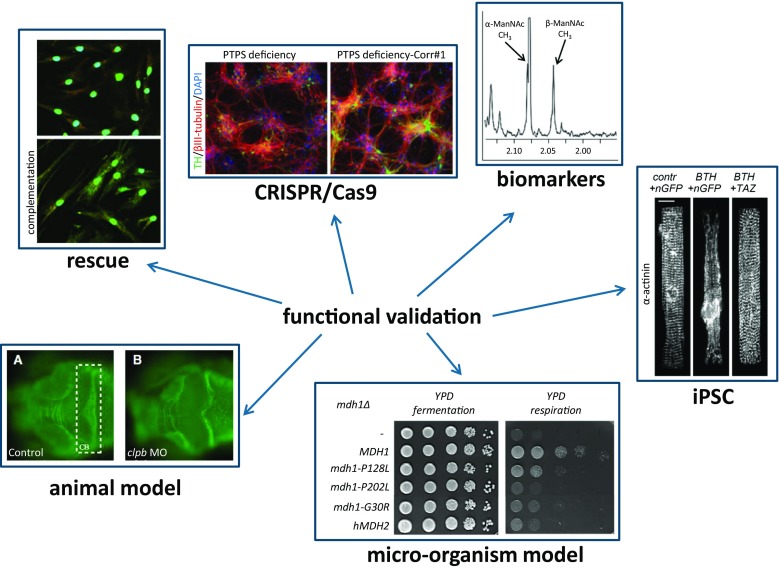
Currently, one of the main challenges in human molecular genetics is the interpretation of rare genetic variants of unknown clinical significance. A conclusive diagnosis is of importance for the patient to obtain certainty about the cause of the disease, for the clinician to be able to provide optimal care to the patient and to predict the disease course, and for the clinical geneticist for genetic counseling of the patient and family members. Conclusive evidence for pathogenicity of genetic variants is therefore crucial. This review gives an introduction to the problem of the interpretation of genetic variants of unknown clinical significance in view of the recent advances in genetic screening, and gives an overview of the possibilities for functional tests that can be performed to answer questions about the function of genes and the functional consequences of genetic variants ("functional genomics") in the field of inborn errors of metabolism (IEM), including several examples of functional genomics studies of mitochondrial disorders and several other IEM.
Conflict of interest statement
R. J. Rodenburg declares that he has no conflict of interest.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
