

RNA secondary structure profiling in zebrafish reveals unique regulatory features
- PMID: 29448945
- PMCID: PMC5815192
- DOI: 10.1186/s12864-018-4497-0
RNA secondary structure profiling in zebrafish reveals unique regulatory features
Abstract
Background: RNA is known to play diverse roles in gene regulation. The clues for this regulatory function of RNA are embedded in its ability to fold into intricate secondary and tertiary structure.
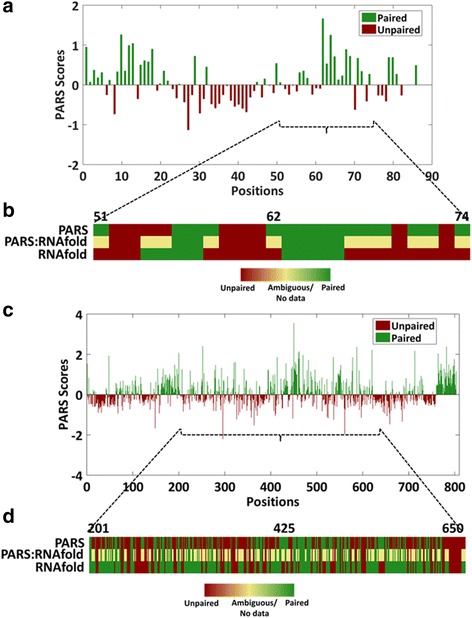
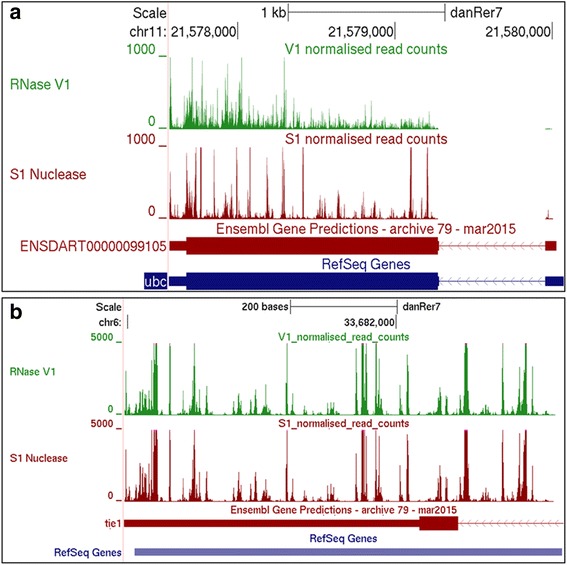
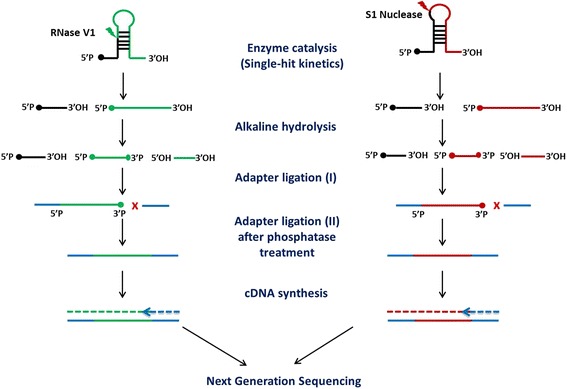
Results: We report the transcriptome-wide RNA secondary structure in zebrafish at single nucleotide resolution using Parallel Analysis of RNA Structure (PARS). This study provides the secondary structure map of zebrafish coding and non-coding RNAs. The single nucleotide pairing probabilities of 54,083 distinct transcripts in the zebrafish genome were documented. We identified RNA secondary structural features embedded in functional units of zebrafish mRNAs. Translation start and stop sites were demarcated by weak structural signals. The coding regions were characterized by the three-nucleotide periodicity of secondary structure and display a codon base specific structural constrain. The splice sites of transcripts were also delineated by distinct signature signals. Relatively higher structural signals were observed at 3' Untranslated Regions (UTRs) compared to Coding DNA Sequence (CDS) and 5' UTRs. The 3' ends of transcripts were also marked by unique structure signals. Secondary structural signals in long non-coding RNAs were also explored to better understand their molecular function.
Conclusions: Our study presents the first PARS-enabled transcriptome-wide secondary structure map of zebrafish, which documents pairing probability of RNA at single nucleotide precision. Our findings open avenues for exploring structural features in zebrafish RNAs and their influence on gene expression.
Keywords: Gene regulation; PARS; RNA secondary structure; Transcriptome; Zebrafish.
Conflict of interest statement
Ethics approval and consent to participate
Fish experiments were performed in strict accordance with the recommendations and guidelines laid down by the CSIR Institute of Genomics and Integrative Biology, India. The protocol was approved by the Institutional Animal Ethics Committee (IAEC) of the CSIR Institute of Genomics and Integrative Biology, India. All efforts were made to minimize animal suffering.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
