

Systematic analysis of protein turnover in primary cells
- PMID: 29449567
- PMCID: PMC5814408
- DOI: 10.1038/s41467-018-03106-1
Systematic analysis of protein turnover in primary cells
Abstract
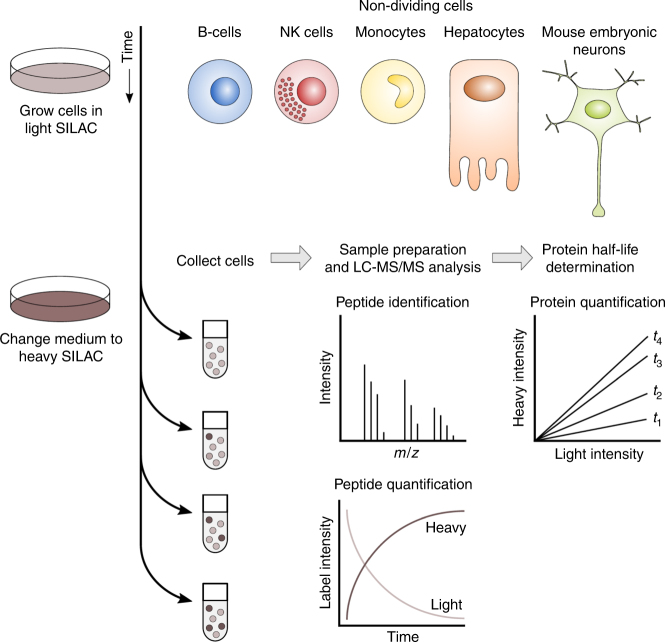
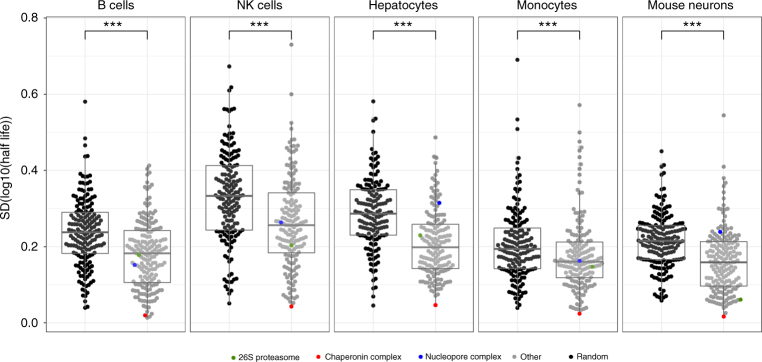
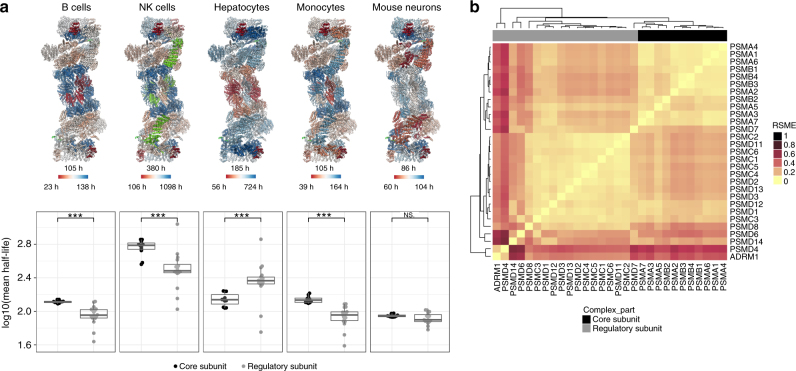
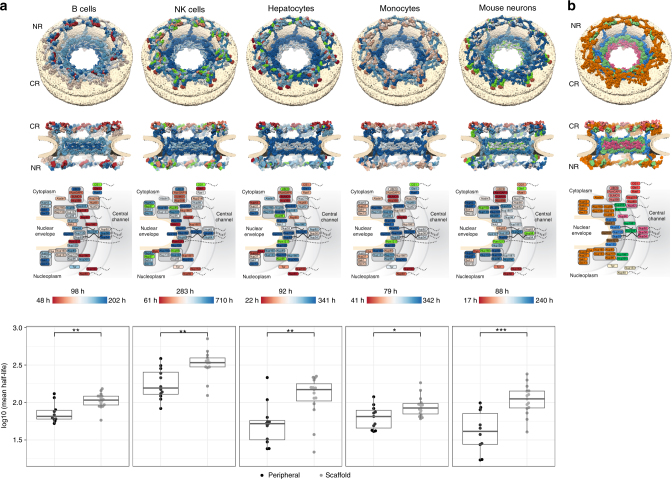
A better understanding of proteostasis in health and disease requires robust methods to determine protein half-lives. Here we improve the precision and accuracy of peptide ion intensity-based quantification, enabling more accurate protein turnover determination in non-dividing cells by dynamic SILAC-based proteomics. This approach allows exact determination of protein half-lives ranging from 10 to >1000 h. We identified 4000-6000 proteins in several non-dividing cell types, corresponding to 9699 unique protein identifications over the entire data set. We observed similar protein half-lives in B-cells, natural killer cells and monocytes, whereas hepatocytes and mouse embryonic neurons show substantial differences. Our data set extends and statistically validates the previous observation that subunits of protein complexes tend to have coherent turnover. Moreover, analysis of different proteasome and nuclear pore complex assemblies suggests that their turnover rate is architecture dependent. These results illustrate that our approach allows investigating protein turnover and its implications in various cell types.
Conflict of interest statement
T.M., G.S., H.F., N.Z., D.P., M.St., G.B. and M.Ba. are employees and/or shareholders of Cellzome GmbH and GlaxoSmithKline. The remaining authors declare no competing financial interests.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
