

Glucocorticoids promote apoptosis of proinflammatory monocytes by inhibiting ERK activity
- PMID: 29449600
- PMCID: PMC5833693
- DOI: 10.1038/s41419-018-0332-4
Glucocorticoids promote apoptosis of proinflammatory monocytes by inhibiting ERK activity
Abstract
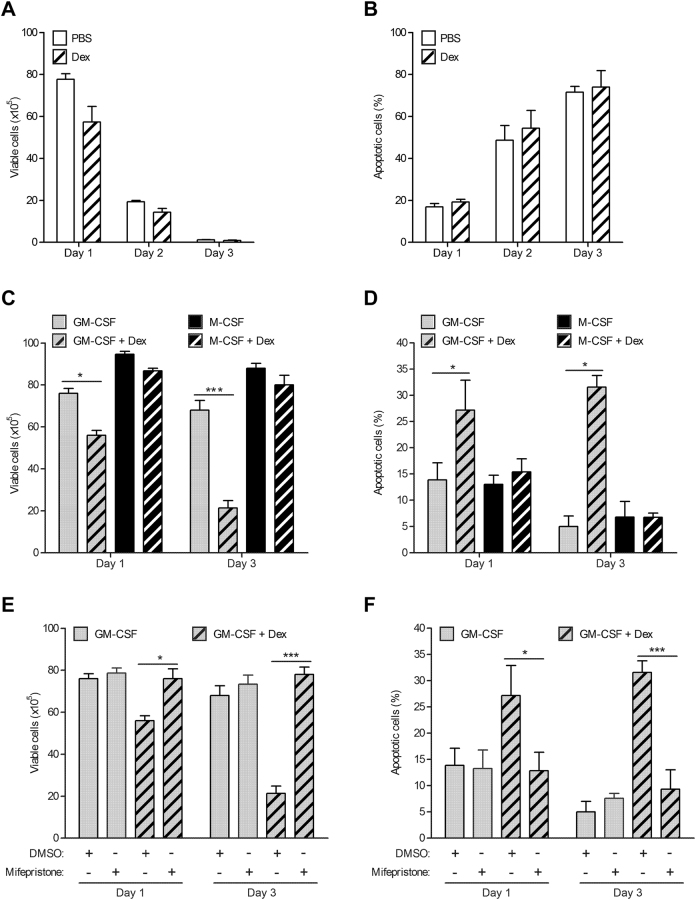
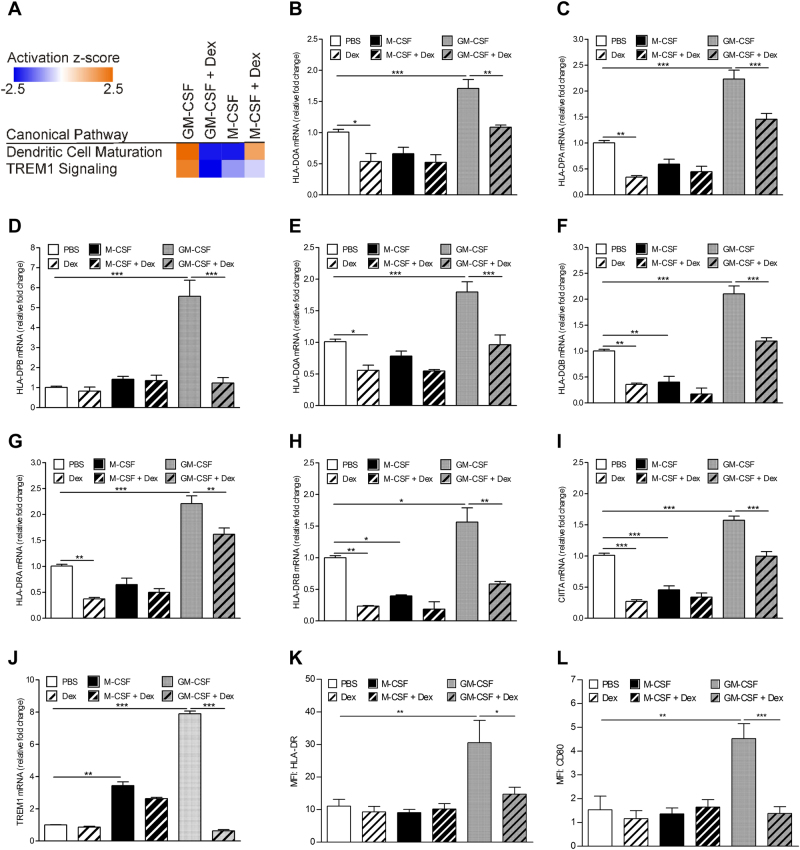
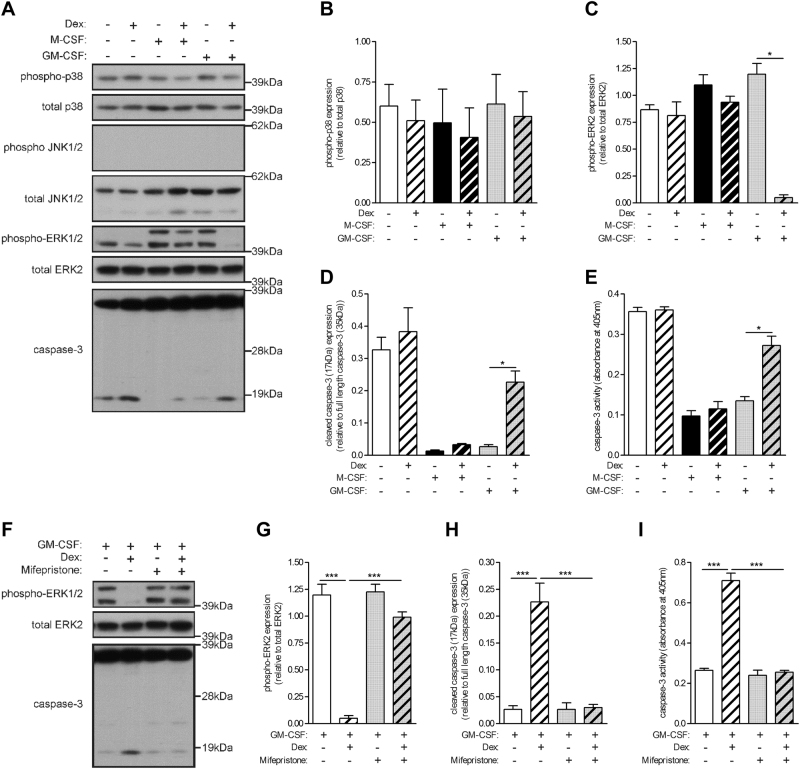
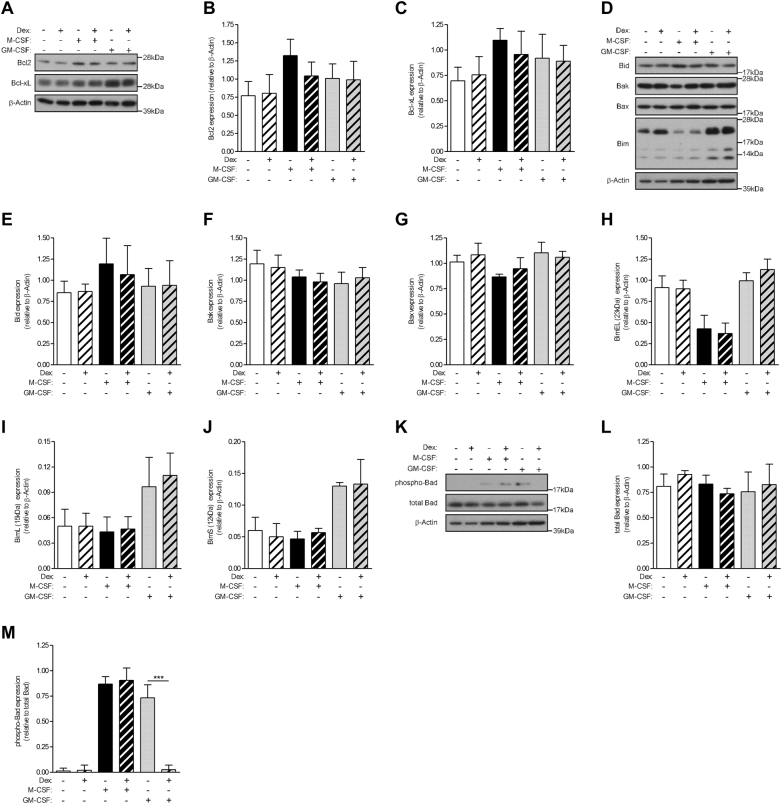
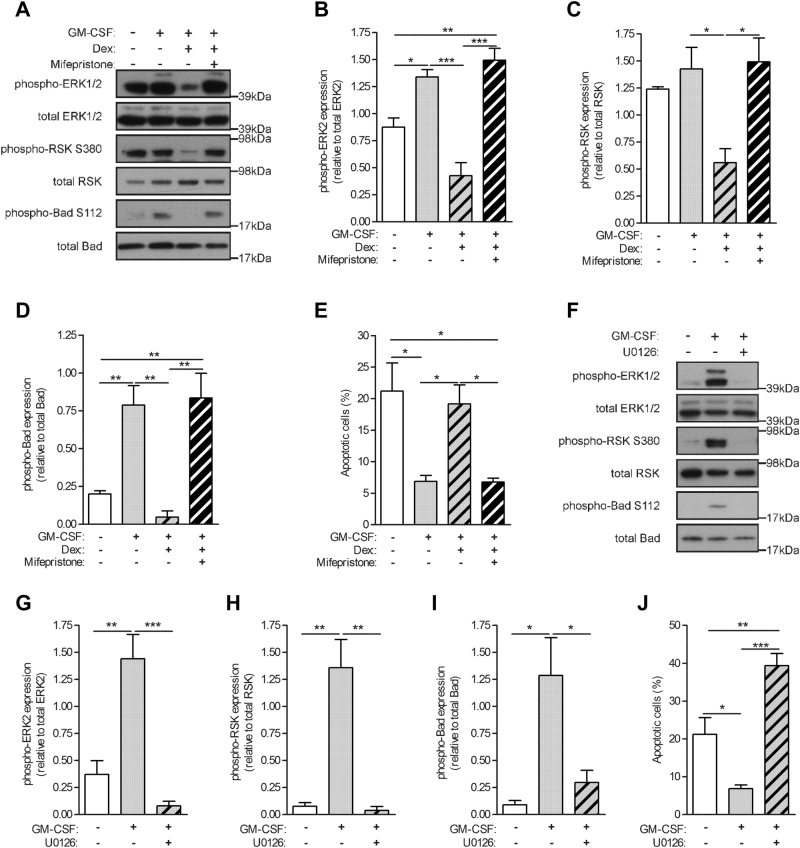
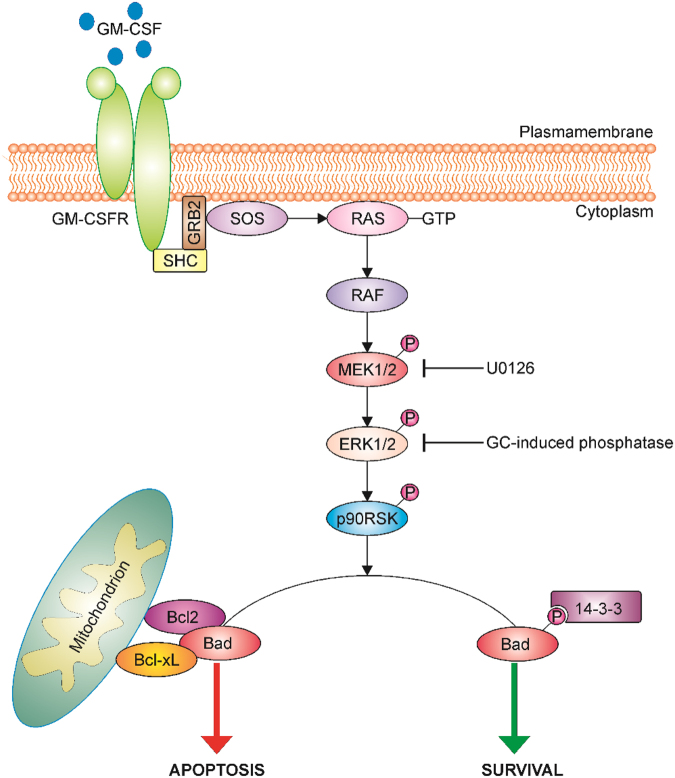
Glucocorticoids (GCs) are potent anti-inflammatory drugs whose mode of action is complex and still debatable. One likely cellular target of GCs are monocytes/macrophages. The role of GCs in monocyte survival is also debated. Although both granulocyte macrophage-colony stimulating factor (GM-CSF) and macrophage-CSF (M-CSF) are important regulators of macrophage lineage functions including their survival, the former is often associated with proinflammatory functions while the latter is important in lineage homeostasis. We report here that the GC, dexamethasone, induces apoptosis in GM-CSF-treated human monocytes while having no impact on M-CSF-induced monocyte survival. To understand how GCs, GM-CSF, and M-CSF are regulating monocyte survival and other functions during inflammation, we firstly examined the transcriptomic changes elicited by these three agents in human monocytes, either acting alone or in combination. Transcriptomic and Ingenuity pathway analyses found that dexamethasone differentially modulated dendritic cell maturation and TREM1 signaling pathways in GM-CSF-treated and M-CSF-treated monocytes, two pathways known to be regulated by ERK1/2 activity. These analyses led us to provide evidence that the GC inhibits ERK1/2 activity selectively in GM-CSF-treated monocytes to induce apoptosis. It is proposed that this inhibition of ERK1/2 activity leads to inactivation of p90 ribosomal-S6 kinase and Bad dephosphorylation leading in turn to enhanced caspase-3 activity and subsequent apoptosis. Furthermore, pharmacological inhibition of GC receptor activity restored the ERK1/2 signaling and prevented the GC-induced apoptosis in GM-CSF-treated monocytes. Increased tissue macrophage numbers, possibly from enhanced survival due to mediators such as GM-CSF, can correlate with inflammatory disease severity; also reduction in these numbers can correlate with the therapeutic benefit of a number of agents, including GCs. We propose that the ERK1/2 signaling pathway promotes survival of GM-CSF-treated proinflammatory monocytes, which can be selectively targeted by GCs as a novel mechanism to reduce local monocyte/macrophage numbers and hence inflammation.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Burgess AW, Metcalf D. The nature and action of granulocyte-macrophage colony stimulating factors. Blood. 1980;56:947–958. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
