

Culture-Facilitated Comparative Genomics of the Facultative Symbiont Hamiltonella defensa
- PMID: 29452355
- PMCID: PMC5841374
- DOI: 10.1093/gbe/evy036
Culture-Facilitated Comparative Genomics of the Facultative Symbiont Hamiltonella defensa
Abstract
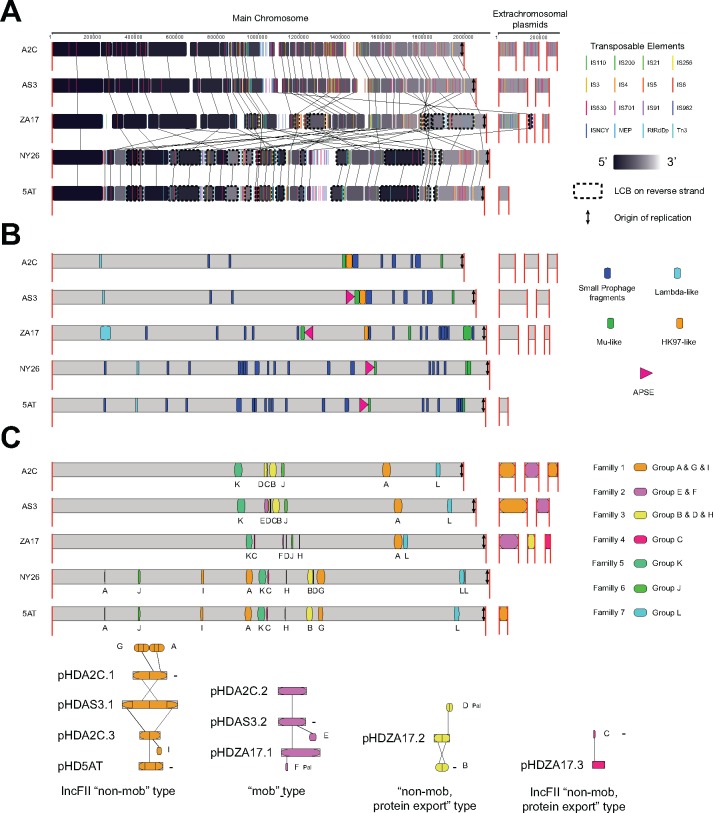
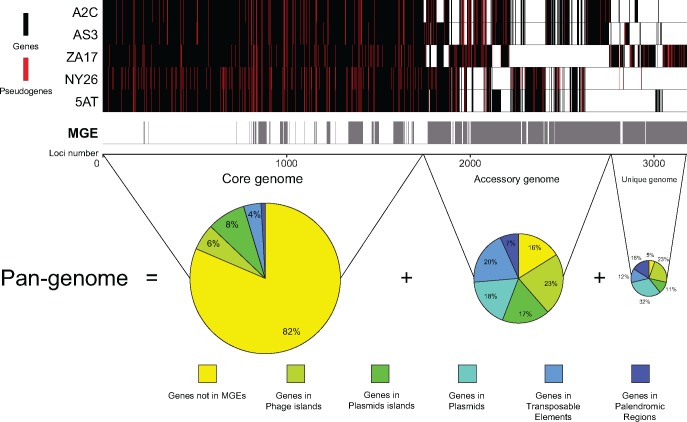
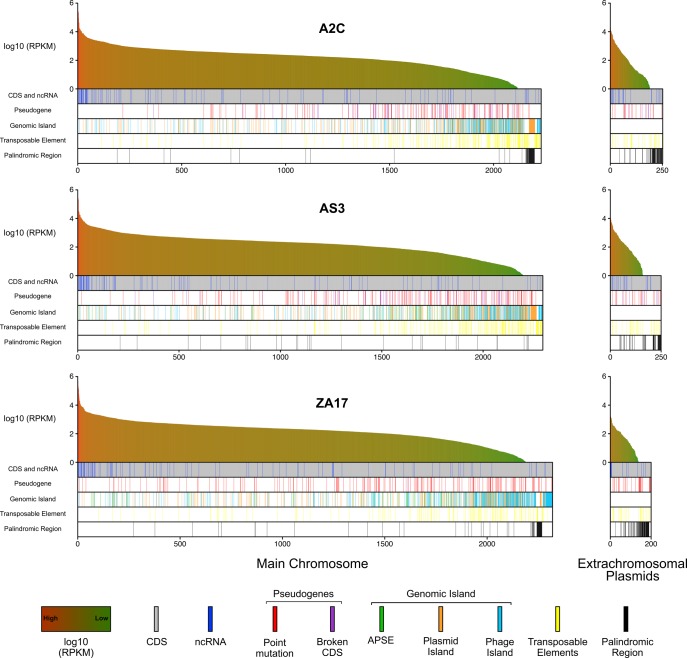
Many insects host facultative, bacterial symbionts that confer conditional fitness benefits to their hosts. Hamiltonella defensa is a common facultative symbiont of aphids that provides protection against parasitoid wasps. Protection levels vary among strains of H. defensa that are also differentially infected by bacteriophages named APSEs. However, little is known about trait variation among strains because only one isolate has been fully sequenced. Generating complete genomes for facultative symbionts is hindered by relatively large genome sizes but low abundances in hosts like aphids that are very small. Here, we took advantage of methods for culturing H. defensa outside of aphids to generate complete genomes and transcriptome data for four strains of H. defensa from the pea aphid Acyrthosiphon pisum. Chosen strains also spanned the breadth of the H. defensa phylogeny and differed in strength of protection conferred against parasitoids. Results indicated that strains shared most genes with roles in nutrient acquisition, metabolism, and essential housekeeping functions. In contrast, the inventory of mobile genetic elements varied substantially, which generated strain specific differences in gene content and genome architecture. In some cases, specific traits correlated with differences in protection against parasitoids, but in others high variation between strains obscured identification of traits with likely roles in defense. Transcriptome data generated continuous distributions to genome assemblies with some genes that were highly expressed and others that were not. Single molecule real-time sequencing further identified differences in DNA methylation patterns and restriction modification systems that provide defense against phage infection.
Figures


References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 2153:403–410. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
