

p66Shc Signaling Mediates Diabetes-Related Cognitive Decline
- PMID: 29453337
- PMCID: PMC5816624
- DOI: 10.1038/s41598-018-21426-6
p66Shc Signaling Mediates Diabetes-Related Cognitive Decline
Abstract
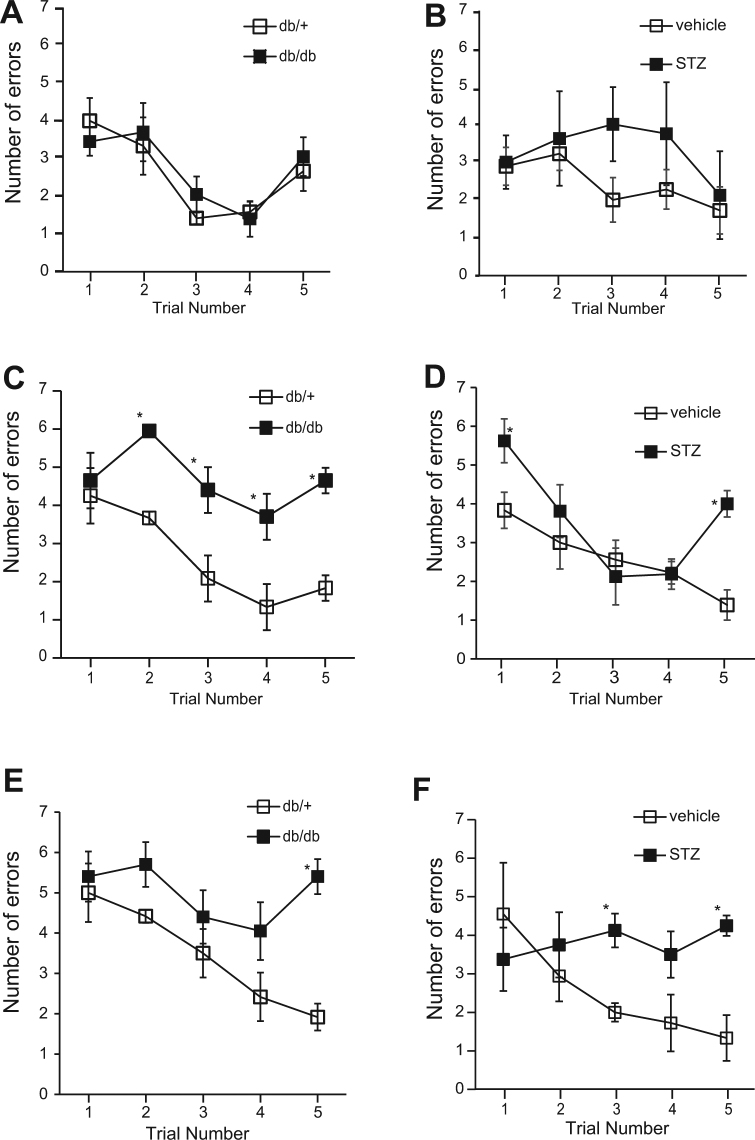
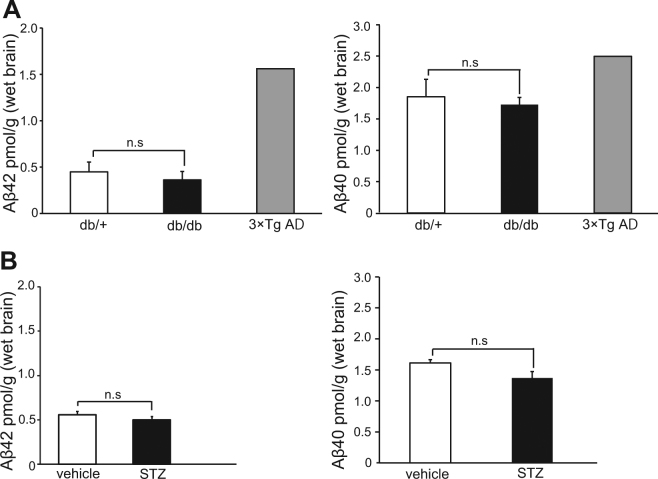
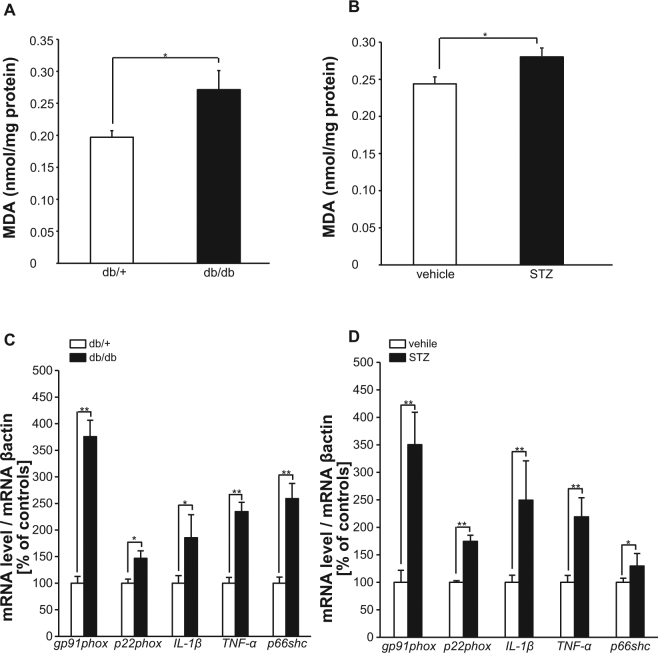
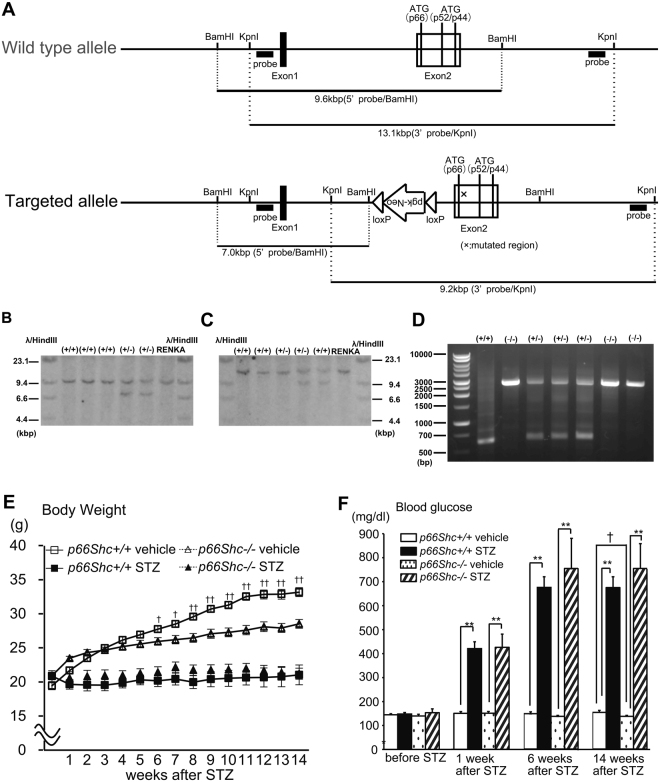
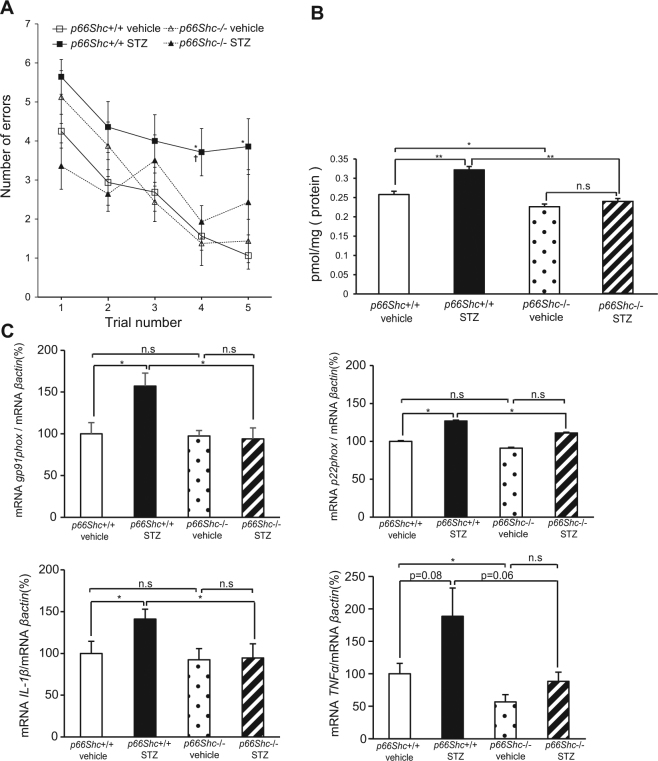
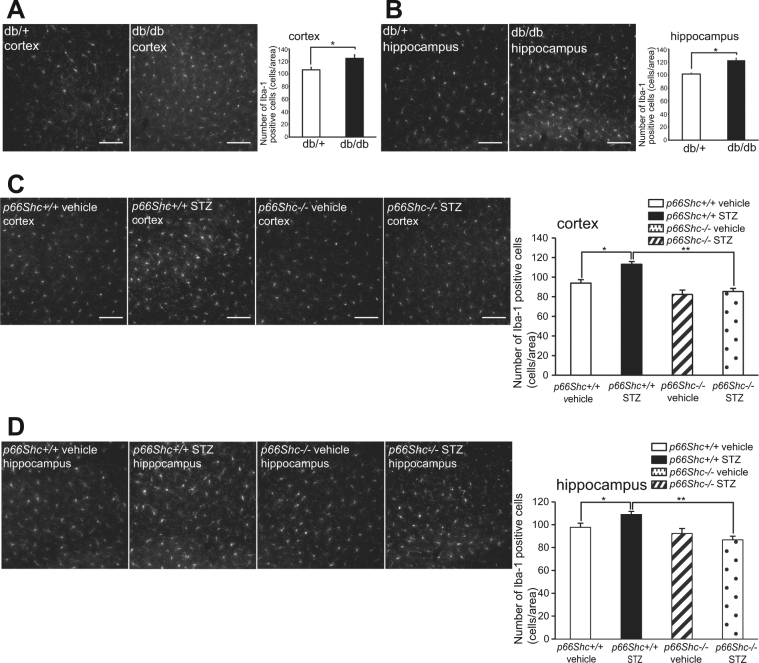
Accumlating evidence have suggested that diabetes mellitus links dementia, notably of Alzheimer's disease (AD). However, the underlying mechanism remains unclear. Several studies have shown oxidative stress (OS) to be one of the major factors in the pathogenesis of diabetic complications. Here we show OS involvement in brain damage in a diabetic animal model that is at least partially mediated through an AD-pathology-independent mechanism apart from amyloid-β accumulation. We investigated the contribution of the p66Shc signaling pathway to diabetes-related cognitive decline using p66Shc knockout (-/-) mice. p66Shc (-/-) mice have less OS in the brain and are resistant to diabetes-induced brain damage. Moreover, p66Shc (-/-) diabetic mice show significantly less cognitive dysfunction and decreased levels of OS and the numbers of microglia. This study postulates a p66Shc-mediated inflammatory cascade leading to OS as a causative pathogenic mechanism in diabetes-associated cognitive impairment that is at least partially mediated through an AD-pathology-independent mechanism.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
