

Homozygous deletion in MYL9 expands the molecular basis of megacystis-microcolon-intestinal hypoperistalsis syndrome
- PMID: 29453416
- PMCID: PMC5945668
- DOI: 10.1038/s41431-017-0055-5
Homozygous deletion in MYL9 expands the molecular basis of megacystis-microcolon-intestinal hypoperistalsis syndrome
Abstract
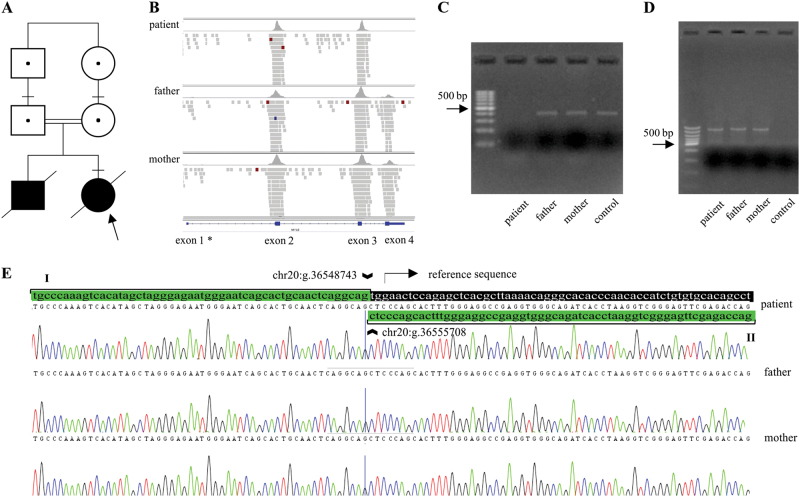
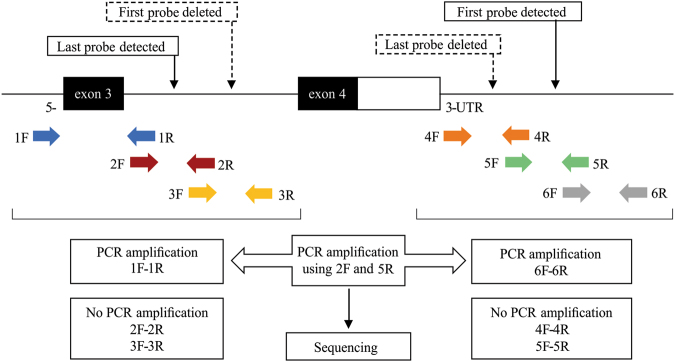
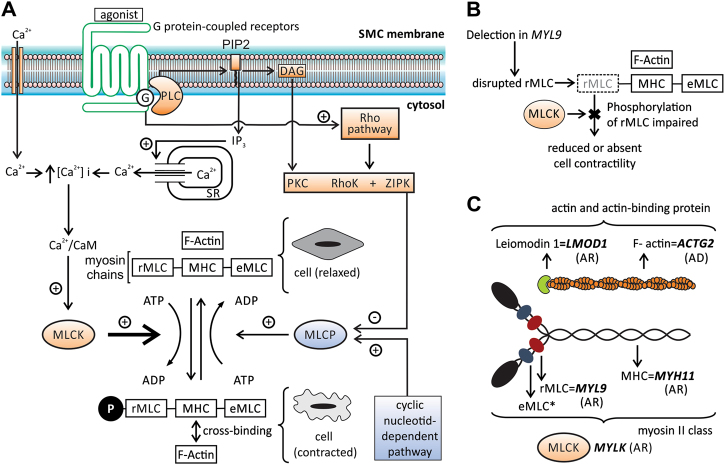
Megacystis-microcolon-intestinal hypoperistalsis syndrome (MMIHS) is a severe disease characterized by functional obstruction in the urinary and gastrointestinal tract. The molecular basis of this condition started to be defined recently, and the genes related to the syndrome (ACTG2-heterozygous variant in sporadic cases; and MYH11 (myosin heavy chain 11), LMOD1 (leiomodin 1) and MYLK (myosin light chain (MLC) kinase)-autosomal recessive inheritance), encode proteins involved in the smooth muscle contraction, supporting a myopathic basis for the disease. In the present article, we described a family with two affected siblings with MMIHS born to consanguineous parents and the molecular investigation performed to define the genetic etiology. Previous whole exome sequencing of the affected child and parents did not identify a candidate gene for the disease in this family, but now we present a reanalysis of the data that led to the identification of a homozygous deletion encompassing the last exon of MYL9 (myosin regulatory light chain 9) in the affected individual. MYL9 gene encodes a regulatory myosin MLC and the phosphorylation of this protein is a crucial step in the contraction process of smooth muscle cell. Despite the absence of human or animal phenotype related to MYL9, a cause-effect relationship between MYL9 and the MMIHS seems biologically plausible. The present study reveals a strong candidate gene for autosomal recessive forms of MMIHS, expanding the molecular basis of this disease and reinforces the myopathic basis of this condition.
Conflict of interest statement
The authors declare that they have no conflict of interest
Figures
Similar articles
-
Compound heterozygous loss of function variants in MYL9 in a child with megacystis-microcolon-intestinal hypoperistalsis syndrome.Mol Genet Genomic Med. 2020 Nov;8(11):e1516. doi: 10.1002/mgg3.1516. Epub 2020 Oct 8. Mol Genet Genomic Med. 2020. PMID: 33031641 Free PMC article.
-
Compound heterozygous variants in MYH11 underlie autosomal recessive megacystis-microcolon-intestinal hypoperistalsis syndrome in a Chinese family.J Hum Genet. 2019 Nov;64(11):1067-1073. doi: 10.1038/s10038-019-0651-z. Epub 2019 Aug 19. J Hum Genet. 2019. PMID: 31427716 Free PMC article.
-
Fetal megacystis-microcolon: Genetic mutational spectrum and identification of PDCL3 as a novel candidate gene.Clin Genet. 2020 Sep;98(3):261-273. doi: 10.1111/cge.13801. Epub 2020 Aug 4. Clin Genet. 2020. PMID: 32621347
-
Consanguinity and its relevance for the incidence of megacystis microcolon intestinal hypoperistalsis syndrome (MMIHS): systematic review.Pediatr Surg Int. 2019 Feb;35(2):175-180. doi: 10.1007/s00383-018-4390-6. Epub 2018 Nov 1. Pediatr Surg Int. 2019. PMID: 30386895
-
Familial megacystis microcolon intestinal hypoperistalsis syndrome: a systematic review.Pediatr Surg Int. 2013 Sep;29(9):947-51. doi: 10.1007/s00383-013-3357-x. Pediatr Surg Int. 2013. PMID: 23955298
Cited by
-
Compound heterozygous loss of function variants in MYL9 in a child with megacystis-microcolon-intestinal hypoperistalsis syndrome.Mol Genet Genomic Med. 2020 Nov;8(11):e1516. doi: 10.1002/mgg3.1516. Epub 2020 Oct 8. Mol Genet Genomic Med. 2020. PMID: 33031641 Free PMC article.
-
A homozygous missense variant in CHRM3 associated with familial urinary bladder disease.Clin Genet. 2019 Dec;96(6):515-520. doi: 10.1111/cge.13631. Epub 2019 Sep 11. Clin Genet. 2019. PMID: 31441039 Free PMC article.
-
Distinct Roles of Smooth Muscle and Non-muscle Myosin Light Chain-Mediated Smooth Muscle Contraction.Front Physiol. 2020 Dec 3;11:593966. doi: 10.3389/fphys.2020.593966. eCollection 2020. Front Physiol. 2020. PMID: 33424621 Free PMC article.
-
Cell-surface proteomic profiling identifies CD72 as a regulator of microglial tiling.bioRxiv [Preprint]. 2025 Jun 5:2025.06.02.657480. doi: 10.1101/2025.06.02.657480. bioRxiv. 2025. PMID: 40501958 Free PMC article. Preprint.
-
Research progress in myosin light chain 9 in malignant tumors.Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2021 Oct 28;46(10):1153-1158. doi: 10.11817/j.issn.1672-7347.2021.200814. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2021. PMID: 34911847 Free PMC article. Chinese, English.
References
-
- Berdon WE, Baker DH, Blanc WA, Gay B, Santulli TV, Donovan C. Megacystis–microcolon–intestinal hypoperistalsis syndrome: a new cause of intestinal obstruction in the newborn. Report of radiologic findings in five newborn girls. Am J Roentgenol. 1976;126:957–64. doi: 10.2214/ajr.126.5.957. - DOI - PubMed
-
- Wangler MF, Gonzaga-Jauregui C, Gambin T, Penney S, Moss T, Chopra A, et al. Heterozygous de novo and inherited mutations in the smooth muscle actin (ACTG2) gene underlie megacystis–microcolon–intestinal hypoperistalsis syndrome. PLoS Genet. 2014;10:e1004258. doi: 10.1371/journal.pgen.1004258. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
