

Neurotrophin-3 restores synaptic plasticity in the striatum of a mouse model of Huntington's disease
- PMID: 29453932
- PMCID: PMC6489824
- DOI: 10.1111/cns.12824
Neurotrophin-3 restores synaptic plasticity in the striatum of a mouse model of Huntington's disease
Abstract
Aims: Neurotrophin-3 (NT-3) is expressed in the mouse striatum; however, it is not clear the NT-3 role in striatal physiology. The expression levels of mRNAs and immune localization of the NT-3 protein and its receptor TrkC are altered in the striatum following damage induced by an in vivo treatment with 3-nitropropionic acid (3-NP), a mitochondrial toxin used to mimic the histopathological hallmarks of Huntington's disease (HD). The aim of this study was to evaluate the role of NT-3 on corticostriatal synaptic transmission and its plasticity in both the control and damaged striatum.
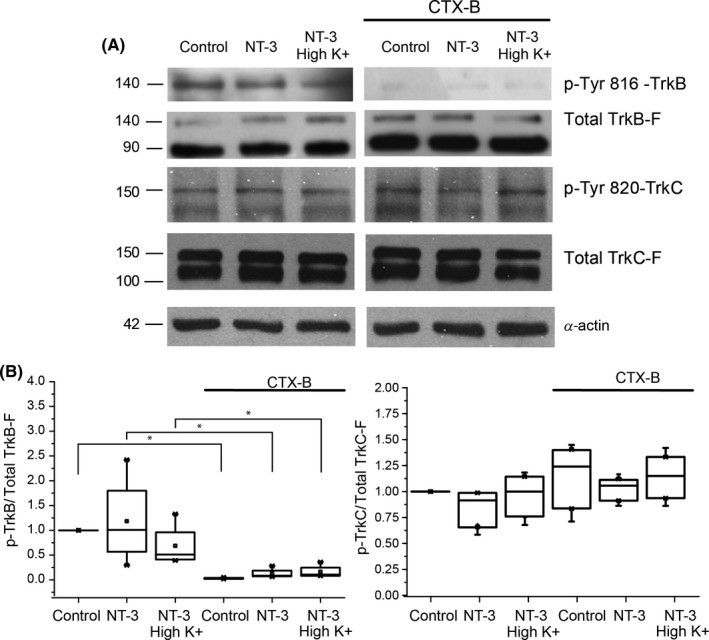
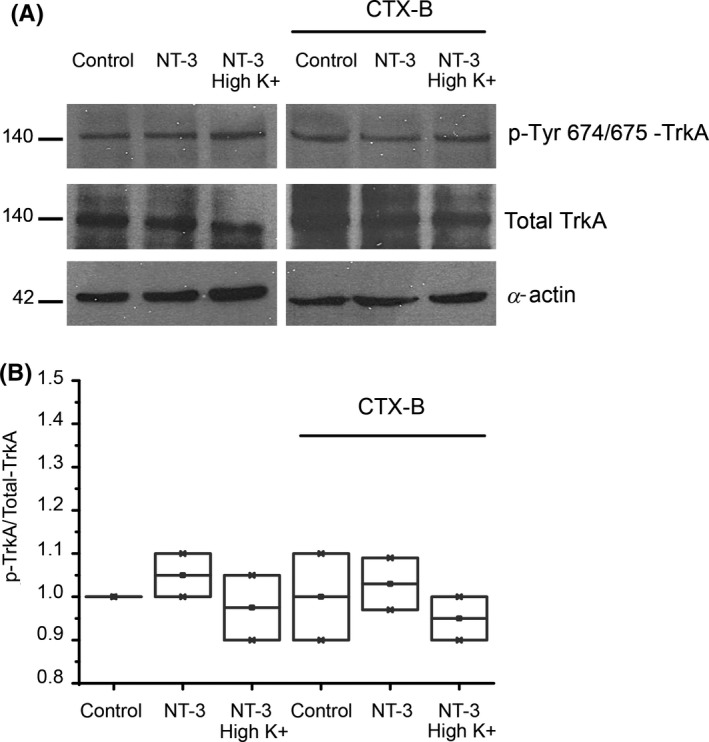
Methods: Corticostriatal population spikes were electrophysiologically recorded and striatal synaptic plasticity was induced by high-frequency stimulation. Further, the phosphorylation status of Trk receptors was tested under conditions that imitated electrophysiological experiments.
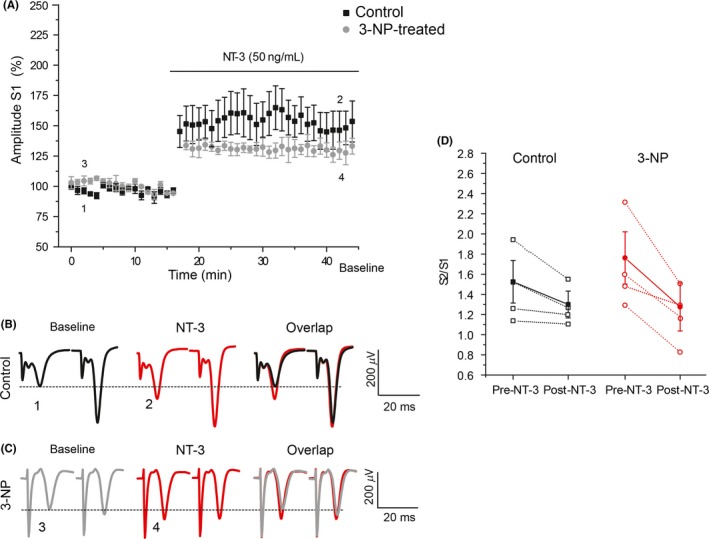
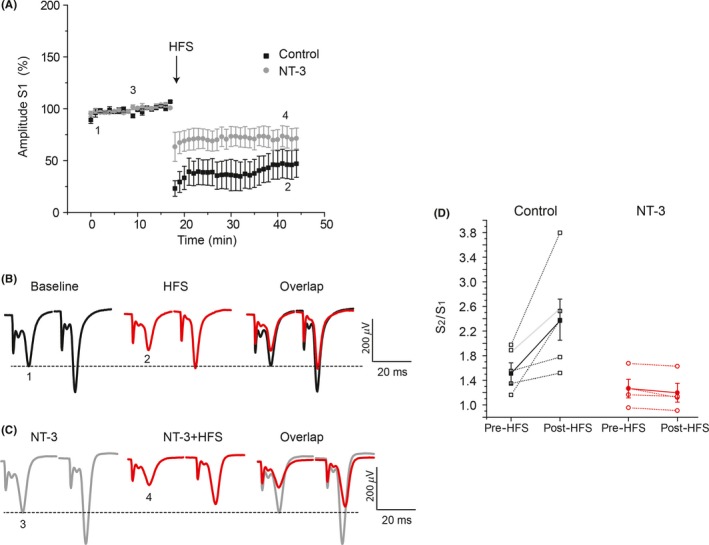
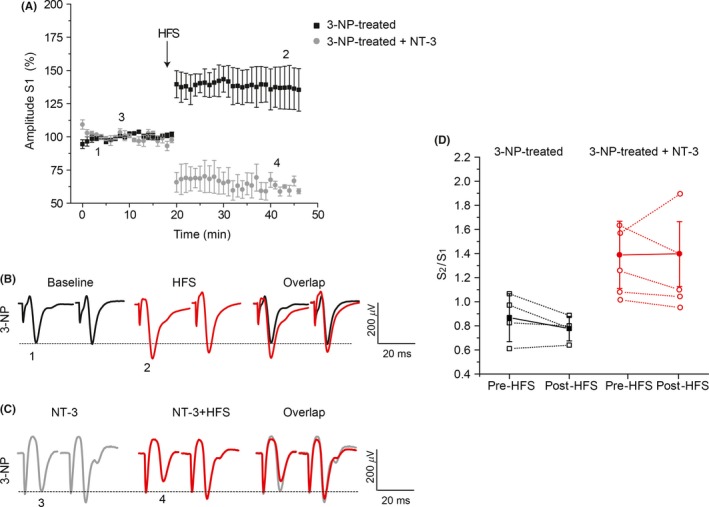
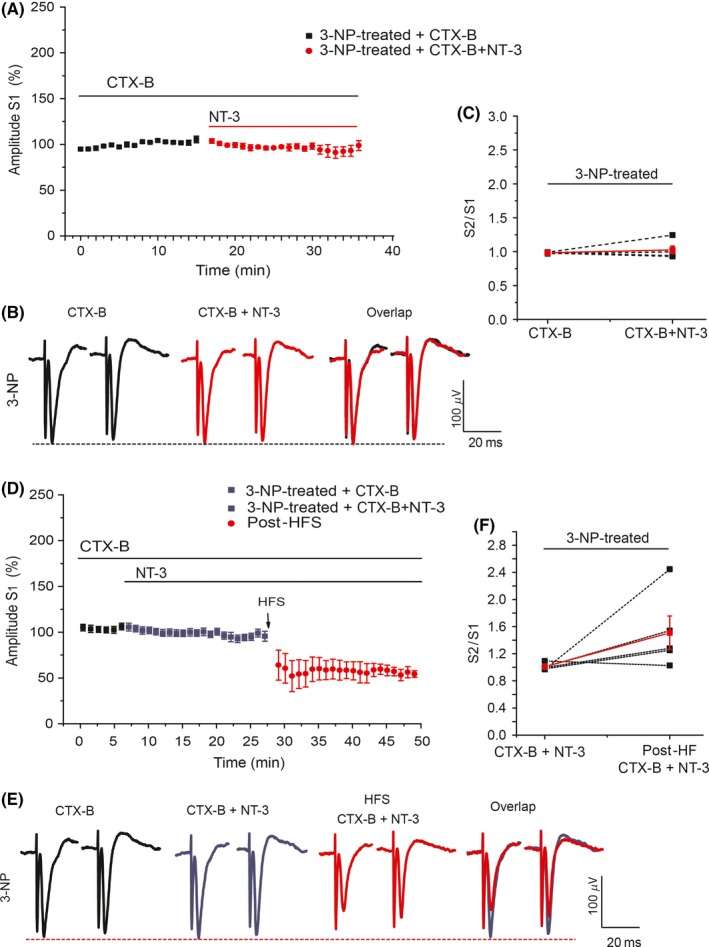
Results: NT-3 modulates both synaptic transmission and plasticity in the striatum; nonetheless, synaptic plasticity was modified by the 3-NP treatment, where instead of producing striatal long-term depression (LTD), long-term potentiation (LTP) was obtained. Moreover, the administration of NT-3 in the recording bath restored the plasticity observed under control conditions (LTD) in this model of striatal degeneration.
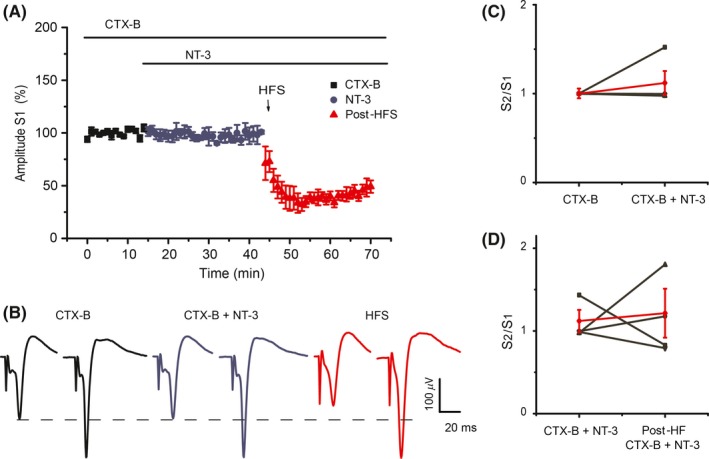
Conclusion: NT-3 modulates corticostriatal transmission through TrkB stimulation and restores striatal LTD by signaling through its TrkC receptor.
Keywords: Huntington's disease; NT-3; TrkC; neurodegeneration; neurotrophins.
© 2018 John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Kremer B, Goldberg P, Andrew SE, et al. A worldwide study of the Huntington's disease mutation. The sensitivity and specificity of measuring CAG repeats. N Engl J Med. 1994;330:1401‐1406. - PubMed
-
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634‐1658. - PubMed
-
- Zuccato C, Ciammola A, Rigamonti D, et al. Loss of huntingtin‐mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493‐498. - PubMed
-
- Urfer R, Tsoulfas P, O'Connell L, Hongo J, Zhao W, Presta LG. High resolution mapping of the binding site of TrkA for nerve growth factor and TrkC for neurotrophin‐3 on the second immunoglobulin‐like domain of the Trk receptors. J Biol Chem. 1998;273:5829‐5840. - PubMed
-
- Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11:272‐280. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
