

Rare Cell Detection by Single-Cell RNA Sequencing as Guided by Single-Molecule RNA FISH
- PMID: 29454938
- PMCID: PMC6078200
- DOI: 10.1016/j.cels.2018.01.014
Rare Cell Detection by Single-Cell RNA Sequencing as Guided by Single-Molecule RNA FISH
Abstract
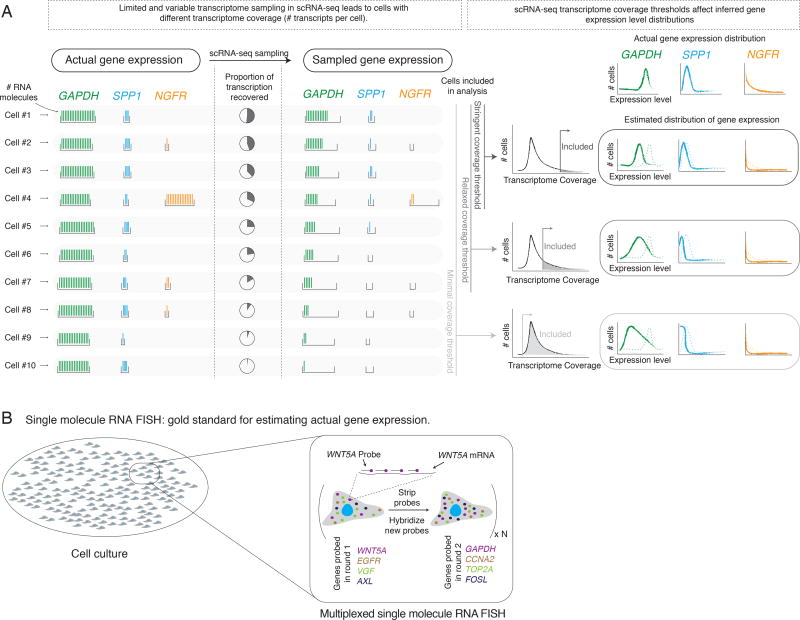
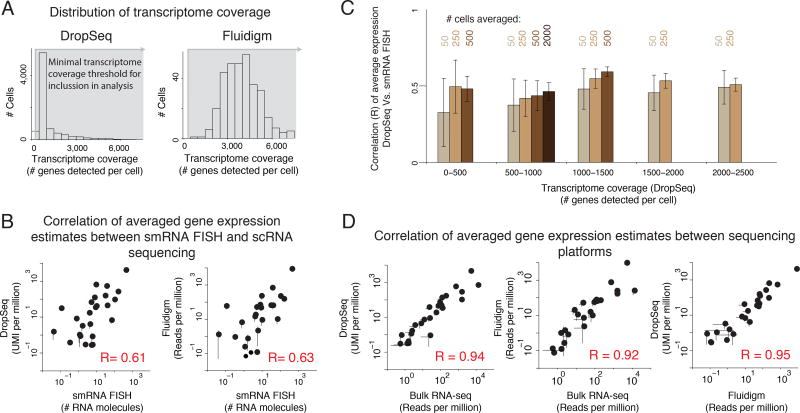
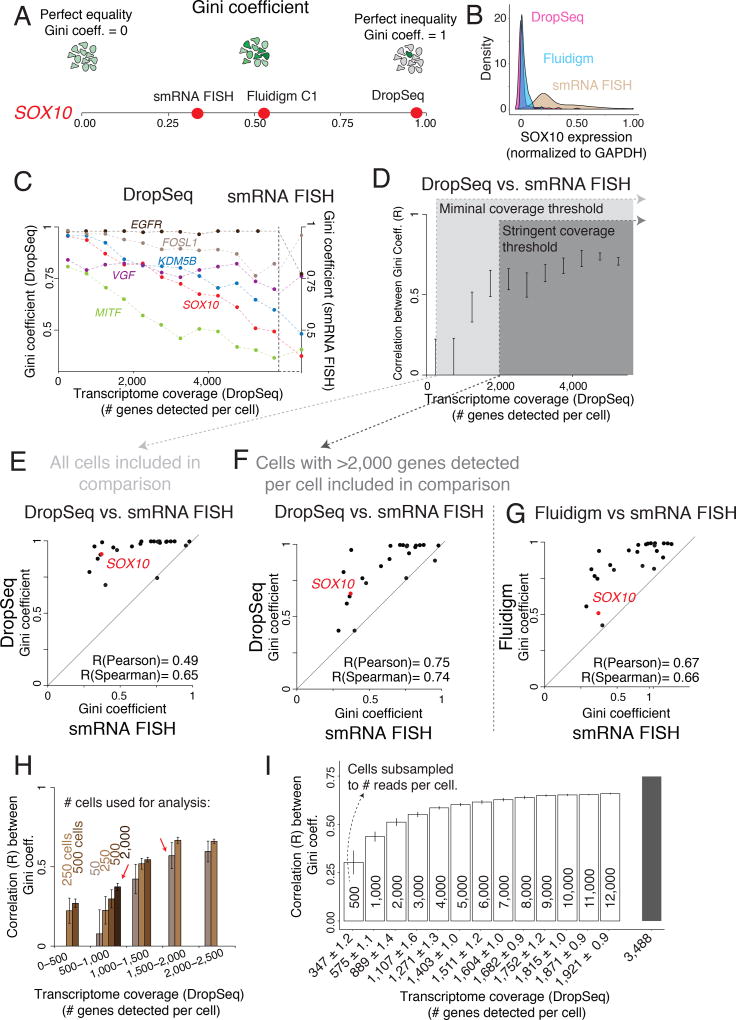
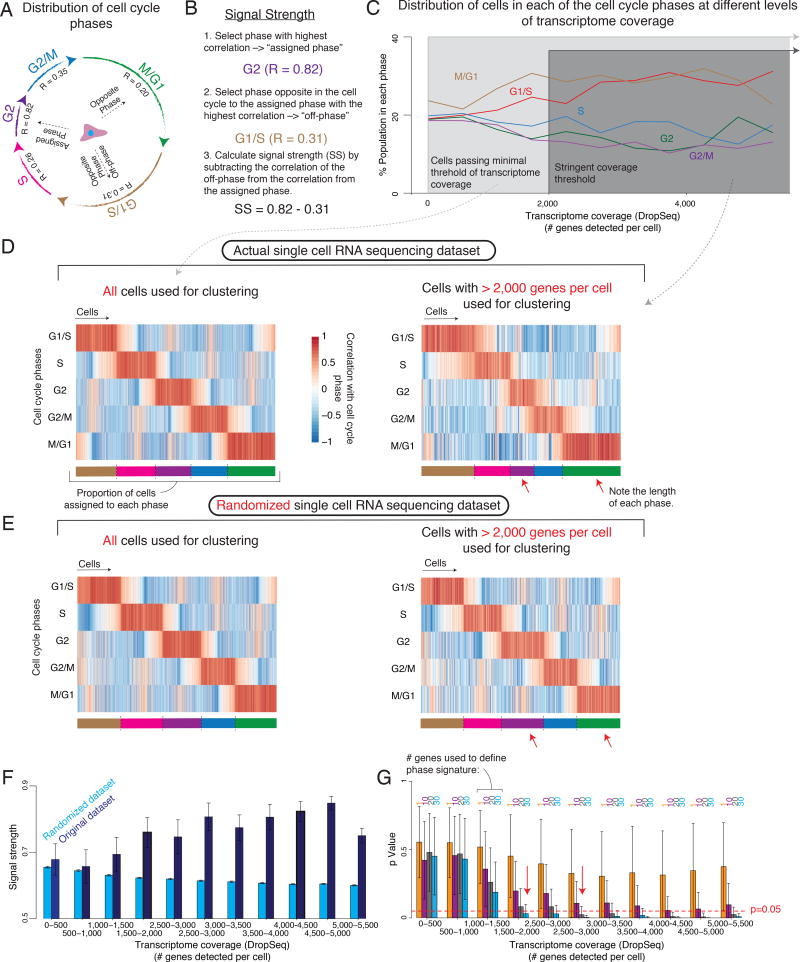
Although single-cell RNA sequencing can reliably detect large-scale transcriptional programs, it is unclear whether it accurately captures the behavior of individual genes, especially those that express only in rare cells. Here, we use single-molecule RNA fluorescence in situ hybridization as a gold standard to assess trade-offs in single-cell RNA-sequencing data for detecting rare cell expression variability. We quantified the gene expression distribution for 26 genes that range from ubiquitous to rarely expressed and found that the correspondence between estimates across platforms improved with both transcriptome coverage and increased number of cells analyzed. Further, by characterizing the trade-off between transcriptome coverage and number of cells analyzed, we show that when the number of genes required to answer a given biological question is small, then greater transcriptome coverage is more important than analyzing large numbers of cells. More generally, our report provides guidelines for selecting quality thresholds for single-cell RNA-sequencing experiments aimed at rare cell analyses.
Keywords: single molecule RNA FISH; single-cell RNA sequencing; single-cell analysis.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
A.R. receives consulting income, and A.R. and S.S. receive royalties related to Stellaris RNA FISH probes. All other authors declare no competing interests.
Figures
Comment in
-
Assessing Inequality in Transcriptomic Data.Cell Syst. 2018 Feb 28;6(2):149-150. doi: 10.1016/j.cels.2018.02.007. Cell Syst. 2018. PMID: 29494801
References
-
- Battich Nico, Stoeger Thomas, Pelkmans Lucas. Control of Transcript Variability in Single Mammalian Cells. Cell. 2015;163(7):1596–1610. - PubMed
-
- Brennecke Philip, Anders Simon, Kim Jong Kyoung, Kołodziejczyk Aleksandra A, Zhang Xiuwei, Proserpio Valentina, Baying Bianka, et al. Accounting for Technical Noise in Single-Cell RNA-Seq Experiments. Nature Methods. 2013;10(11):1093–95. - PubMed
-
- Cabili Moran N, Dunagin Margaret C, McClanahan Patrick D, Biaesch Andrew, Padovan-Merhar Olivia, Regev Aviv, Rinn John L, Raj Arjun. Genome Biology. 1. Vol. 16. BioMed Central Ltd; 2015. Localization and Abundance Analysis of Human lncRNAs at Single-Cell and Single-Molecule Resolution; p. 20. - PMC - PubMed
-
- Dijk David van, Nainys Juozas, Sharma Roshan, Kathail Pooja, Carr Ambrose J, Moon Kevin R, Mazutis Linas, Wolf Guy, Krishnaswamy Smita, Pe’er Dana. MAGIC: A Diffusion-Based Imputation Method Reveals Gene-Gene Interactions in Single-Cell RNA-Sequencing Data. bioRxiv. 2017 doi: 10.1101/111591. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
