

Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities
- PMID: 29455668
- PMCID: PMC5817860
- DOI: 10.1186/s12943-018-0796-y
Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities
Abstract
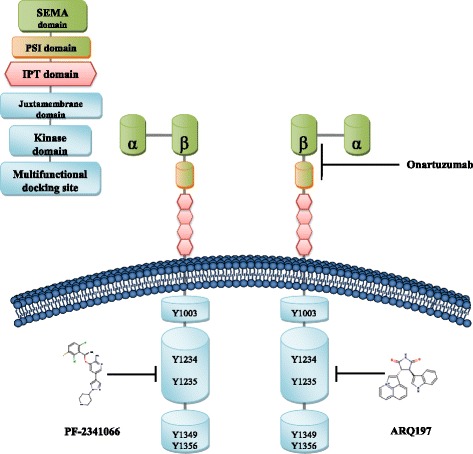
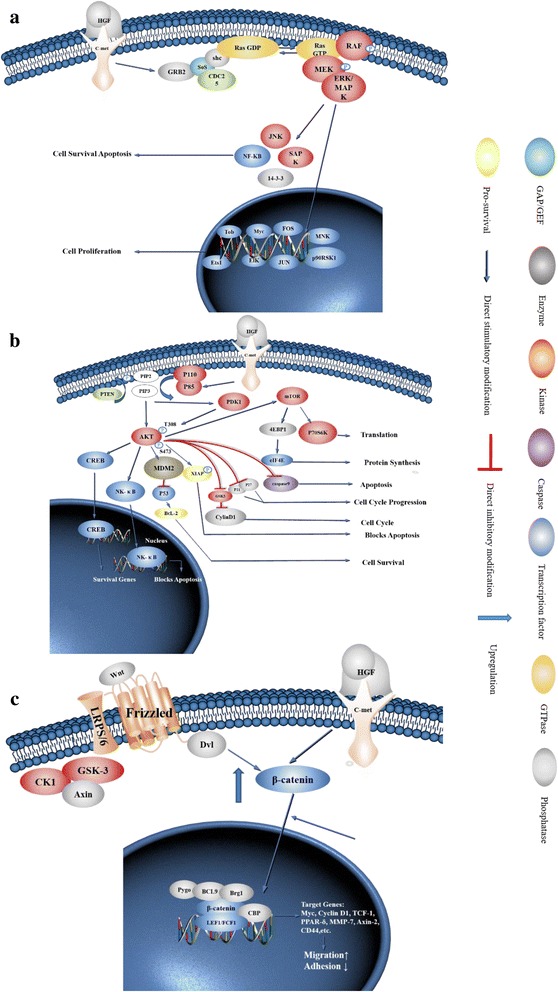
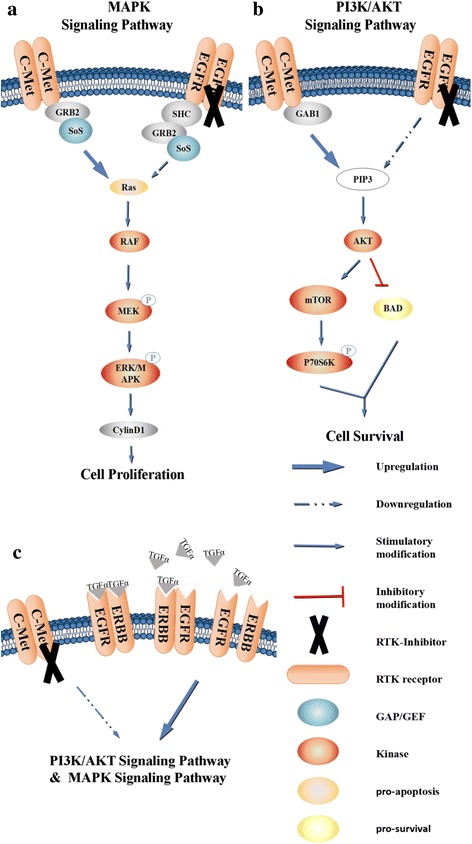
c-Met is a receptor tyrosine kinase belonging to the MET (MNNG HOS transforming gene) family, and is expressed on the surfaces of various cells. Hepatocyte growth factor (HGF) is the ligand for this receptor. The binding of HGF to c-Met initiates a series of intracellular signals that mediate embryogenesis and wound healing in normal cells. However, in cancer cells, aberrant HGF/c-Met axis activation, which is closely related to c-Met gene mutations, overexpression, and amplification, promotes tumor development and progression by stimulating the PI3K/AKT, Ras/MAPK, JAK/STAT, SRC, Wnt/β-catenin, and other signaling pathways. Thus, c-Met and its associated signaling pathways are clinically important therapeutic targets. In this review, we elaborate on the molecular structure of c-Met and HGF and the mechanism through which their interaction activates the PI3K/AKT, Ras/MAPK, and Wnt signaling pathways. We also summarize the connection between c-Met and RON and EGFR, which are also receptor tyrosine kinases. Finally, we introduce the current therapeutic drugs that target c-Met in primary tumors, and their use in clinical research.
Keywords: EGFR; HGF/c-Met; PI3K/AKT; RON; Ras/MAPK; Therapeutic strategy; Wnt.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
