

Heterozygous Mutations in OAS1 Cause Infantile-Onset Pulmonary Alveolar Proteinosis with Hypogammaglobulinemia
- PMID: 29455859
- PMCID: PMC5985284
- DOI: 10.1016/j.ajhg.2018.01.019
Heterozygous Mutations in OAS1 Cause Infantile-Onset Pulmonary Alveolar Proteinosis with Hypogammaglobulinemia
Abstract
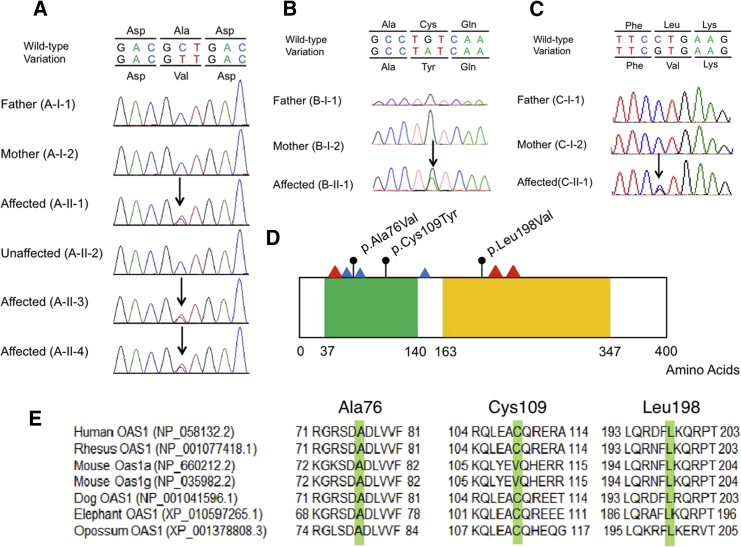
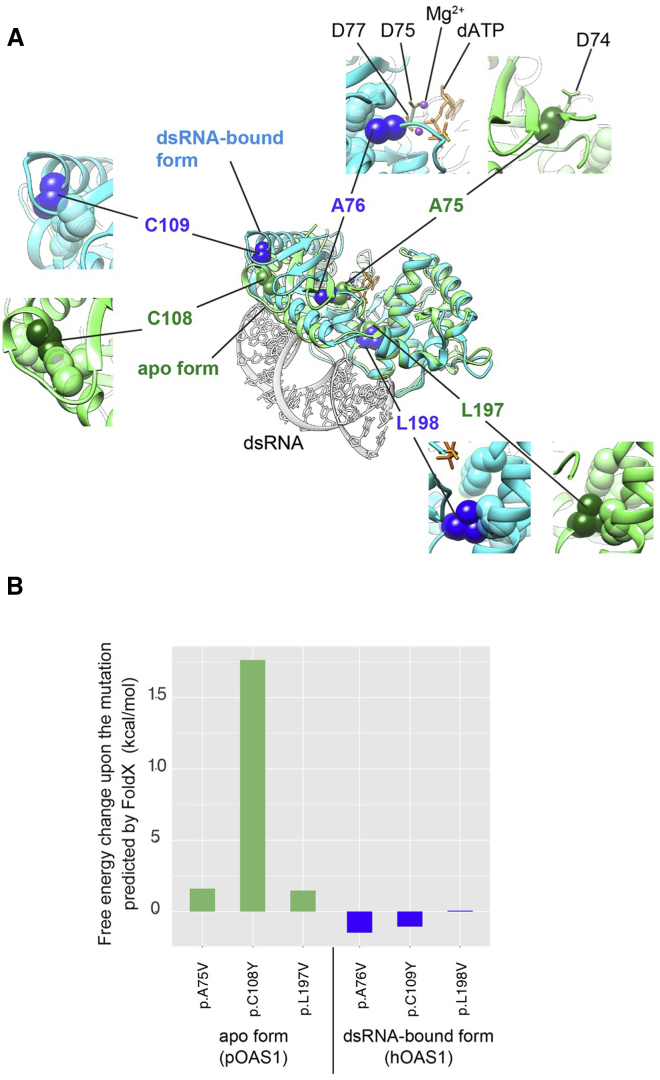
Pulmonary alveolar proteinosis (PAP) is characterized by accumulation of a surfactant-like substance in alveolar spaces and hypoxemic respiratory failure. Genetic PAP (GPAP) is caused by mutations in genes encoding surfactant proteins or genes encoding a surfactant phospholipid transporter in alveolar type II epithelial cells. GPAP is also caused by mutations in genes whose products are implicated in surfactant catabolism in alveolar macrophages (AMs). We performed whole-exome sequence analysis in a family affected by infantile-onset PAP with hypogammaglobulinemia without causative mutations in genes associated with PAP: SFTPB, SFTPC, ABCA3, CSF2RA, CSF2RB, and GATA2. We identified a heterozygous missense variation in OAS1, encoding 2,'5'-oligoadenylate synthetase 1 (OAS1) in three affected siblings, but not in unaffected family members. Deep sequence analysis with next-generation sequencing indicated 3.81% mosaicism of this variant in DNA from their mother's peripheral blood leukocytes, suggesting that PAP observed in this family could be inherited as an autosomal-dominant trait from the mother. We identified two additional de novo heterozygous missense variations of OAS1 in two unrelated simplex individuals also manifesting infantile-onset PAP with hypogammaglobulinemia. PAP in the two simplex individuals resolved after hematopoietic stem cell transplantation, indicating that OAS1 dysfunction is associated with impaired surfactant catabolism due to the defects in AMs.
Keywords: 2′,5′-oligoadenylate synthetase 1; OAS1; PAP; alveolar macrophage; hypogammaglobulinemia; pulmonary alveolar proteinosis.
Copyright © 2018 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Pulmonary alveolar proteinosis due to heterozygous mutation in OAS1: Whole lung lavages for long-term bridging to hematopoietic stem cell transplantation.Pediatr Pulmonol. 2022 Jan;57(1):273-277. doi: 10.1002/ppul.25728. Epub 2021 Nov 15. Pediatr Pulmonol. 2022. PMID: 34647697
-
Mutation of SFTPC in infantile pulmonary alveolar proteinosis with or without fibrosing lung disease.Am J Med Genet A. 2004 Apr 1;126A(1):18-26. doi: 10.1002/ajmg.a.20670. Am J Med Genet A. 2004. PMID: 15039969
-
A murine model of hereditary pulmonary alveolar proteinosis caused by homozygous Csf2ra gene disruption.Am J Physiol Lung Cell Mol Physiol. 2022 Mar 1;322(3):L438-L448. doi: 10.1152/ajplung.00175.2021. Epub 2022 Jan 19. Am J Physiol Lung Cell Mol Physiol. 2022. PMID: 35043685 Free PMC article.
-
Pulmonary alveolar proteinosis.Nat Rev Dis Primers. 2019 Mar 7;5(1):16. doi: 10.1038/s41572-019-0066-3. Nat Rev Dis Primers. 2019. PMID: 30846703 Review.
-
Pulmonary Alveolar Proteinosis Syndrome.Clin Chest Med. 2016 Sep;37(3):431-40. doi: 10.1016/j.ccm.2016.04.006. Epub 2016 Jun 17. Clin Chest Med. 2016. PMID: 27514590 Free PMC article. Review.
Cited by
-
Heterozygous OAS1 gain-of-function variants cause an autoinflammatory immunodeficiency.Sci Immunol. 2021 Jun 18;6(60):eabf9564. doi: 10.1126/sciimmunol.abf9564. Sci Immunol. 2021. PMID: 34145065 Free PMC article.
-
Alveolar proteinosis of genetic origins.Eur Respir Rev. 2020 Oct 28;29(158):190187. doi: 10.1183/16000617.0187-2019. Print 2020 Dec 31. Eur Respir Rev. 2020. PMID: 33115790 Free PMC article.
-
Natural Autoantibodies in Chronic Pulmonary Diseases.Int J Mol Sci. 2020 Feb 8;21(3):1138. doi: 10.3390/ijms21031138. Int J Mol Sci. 2020. PMID: 32046322 Free PMC article. Review.
-
Interstitial Lung Disease in Immunocompromised Children.Diagnostics (Basel). 2022 Dec 26;13(1):64. doi: 10.3390/diagnostics13010064. Diagnostics (Basel). 2022. PMID: 36611354 Free PMC article.
-
A de novo dominant-negative variant is associated with OTULIN-related autoinflammatory syndrome.J Exp Med. 2024 Jun 3;221(6):e20231941. doi: 10.1084/jem.20231941. Epub 2024 Apr 23. J Exp Med. 2024. PMID: 38652464 Free PMC article.
References
-
- Jobe A.H. Pulmonary surfactant therapy. N. Engl. J. Med. 1993;328:861–868. - PubMed
-
- Whitsett J.A., Weaver T.E. Hydrophobic surfactant proteins in lung function and disease. N. Engl. J. Med. 2002;347:2141–2148. - PubMed
-
- Fitzgerald M.L., Xavier R., Haley K.J., Welti R., Goss J.L., Brown C.E., Zhuang D.Z., Bell S.A., Lu N., McKee M. ABCA3 inactivation in mice causes respiratory failure, loss of pulmonary surfactant, and depletion of lung phosphatidylglycerol. J. Lipid Res. 2007;48:621–632. - PubMed
-
- Uchida K., Nakata K., Trapnell B.C., Terakawa T., Hamano E., Mikami A., Matsushita I., Seymour J.F., Oh-Eda M., Ishige I. High-affinity autoantibodies specifically eliminate granulocyte-macrophage colony-stimulating factor activity in the lungs of patients with idiopathic pulmonary alveolar proteinosis. Blood. 2004;103:1089–1098. - PubMed
-
- Trapnell B.C., Whitsett J.A., Nakata K. Pulmonary alveolar proteinosis. N. Engl. J. Med. 2003;349:2527–2539. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
