

Recon3D enables a three-dimensional view of gene variation in human metabolism
- PMID: 29457794
- PMCID: PMC5840010
- DOI: 10.1038/nbt.4072
Recon3D enables a three-dimensional view of gene variation in human metabolism
Abstract
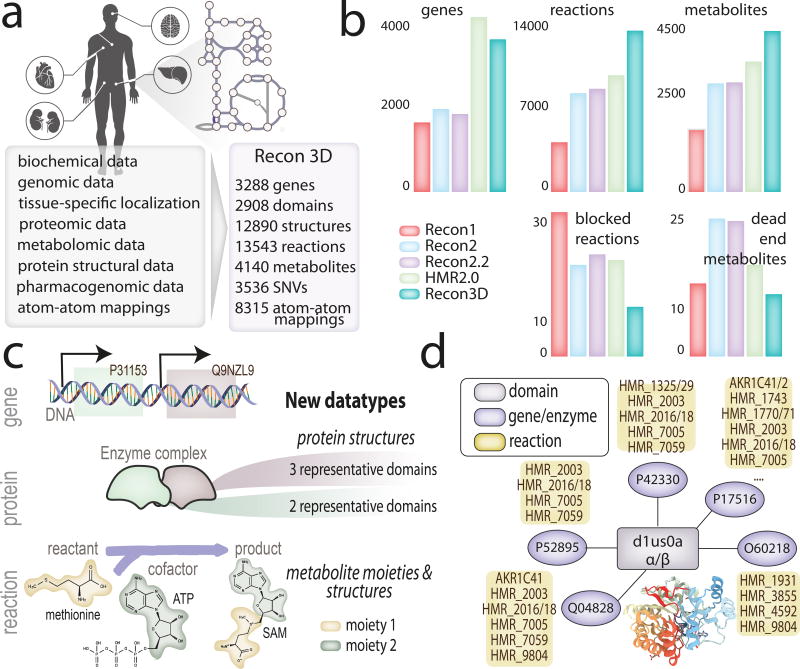
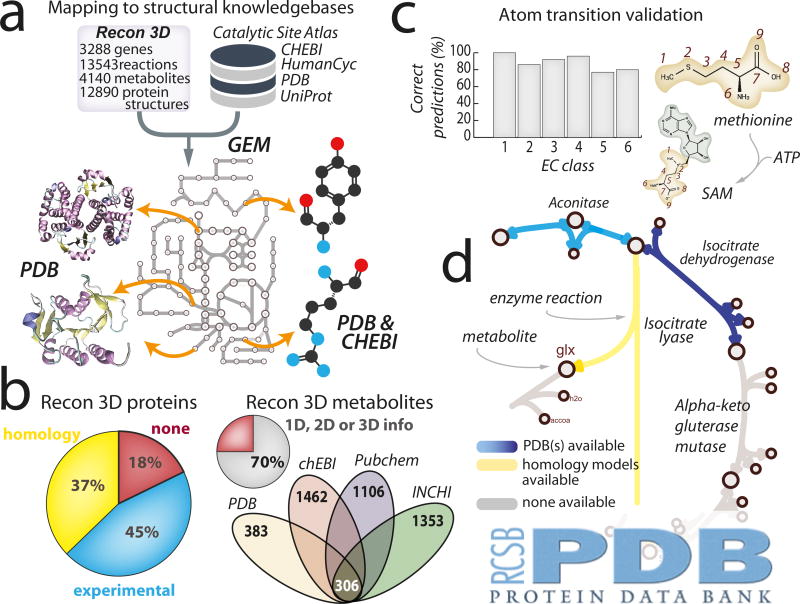
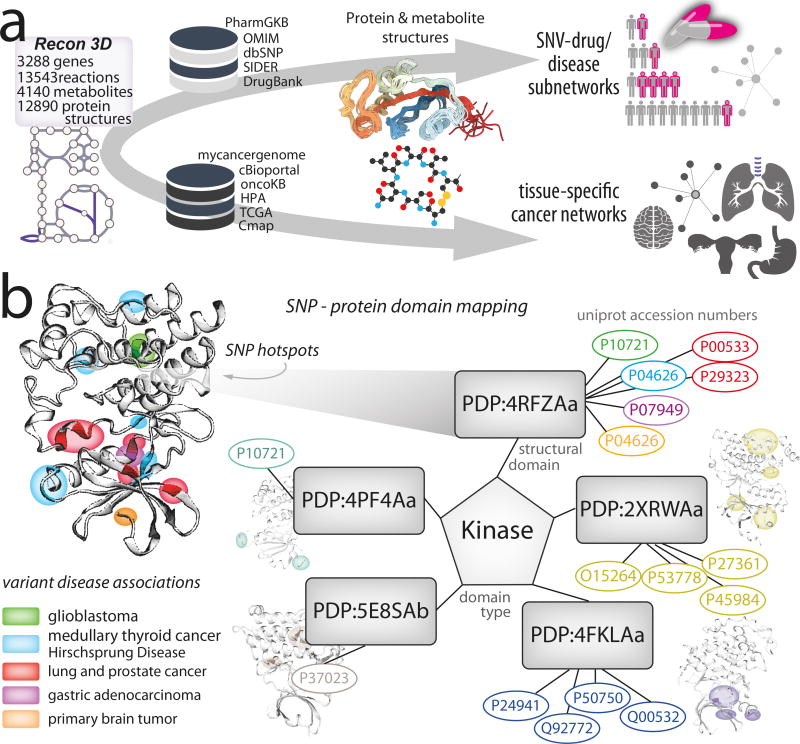
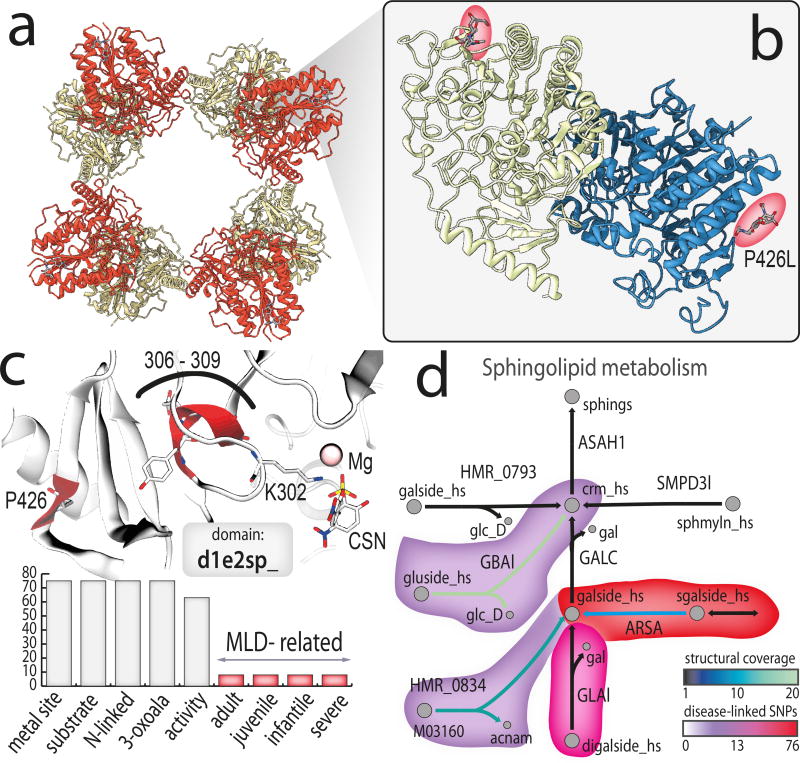
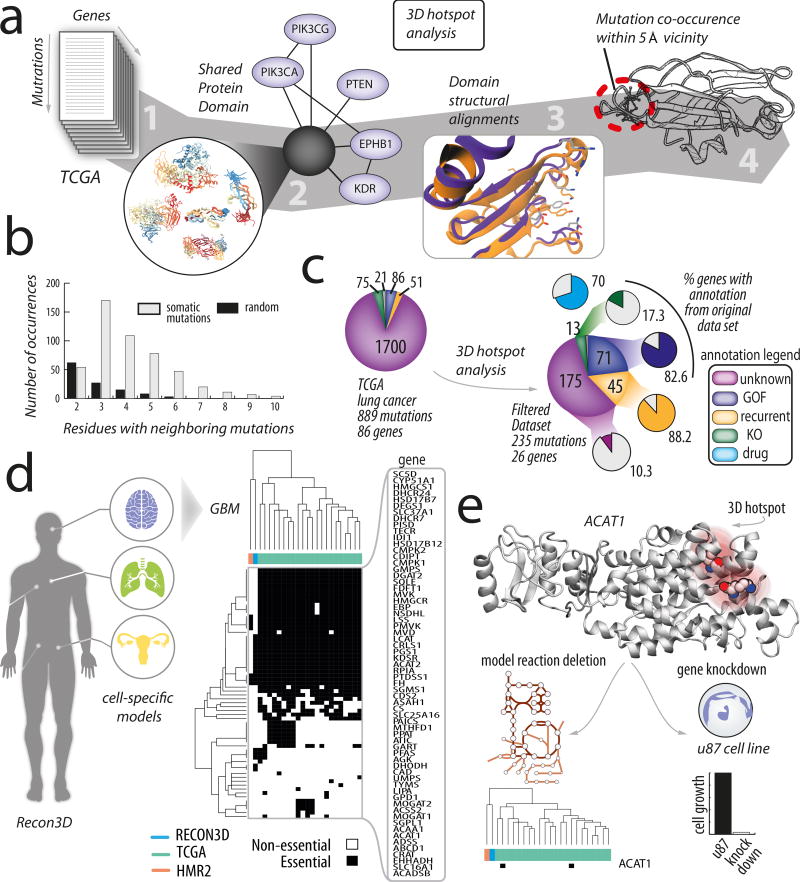
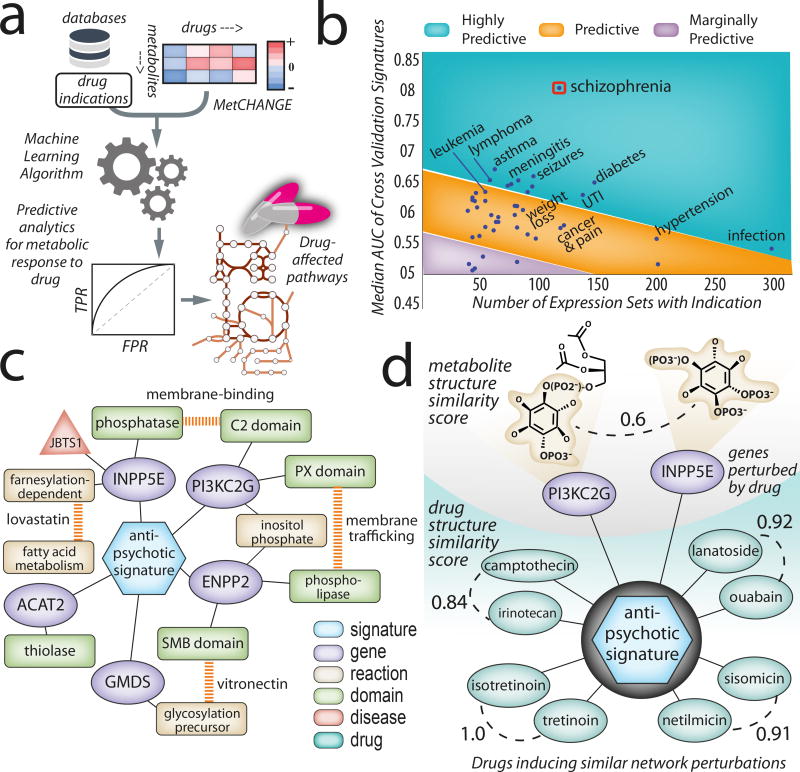
Genome-scale network reconstructions have helped uncover the molecular basis of metabolism. Here we present Recon3D, a computational resource that includes three-dimensional (3D) metabolite and protein structure data and enables integrated analyses of metabolic functions in humans. We use Recon3D to functionally characterize mutations associated with disease, and identify metabolic response signatures that are caused by exposure to certain drugs. Recon3D represents the most comprehensive human metabolic network model to date, accounting for 3,288 open reading frames (representing 17% of functionally annotated human genes), 13,543 metabolic reactions involving 4,140 unique metabolites, and 12,890 protein structures. These data provide a unique resource for investigating molecular mechanisms of human metabolism. Recon3D is available at http://vmh.life.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
