

The genomic underpinnings of eukaryotic virus taxonomy: creating a sequence-based framework for family-level virus classification
- PMID: 29458427
- PMCID: PMC5819261
- DOI: 10.1186/s40168-018-0422-7
The genomic underpinnings of eukaryotic virus taxonomy: creating a sequence-based framework for family-level virus classification
Abstract
Background: The International Committee on Taxonomy of Viruses (ICTV) classifies viruses into families, genera and species and provides a regulated system for their nomenclature that is universally used in virus descriptions. Virus taxonomic assignments have traditionally been based upon virus phenotypic properties such as host range, virion morphology and replication mechanisms, particularly at family level. However, gene sequence comparisons provide a clearer guide to their evolutionary relationships and provide the only information that may guide the incorporation of viruses detected in environmental (metagenomic) studies that lack any phenotypic data.
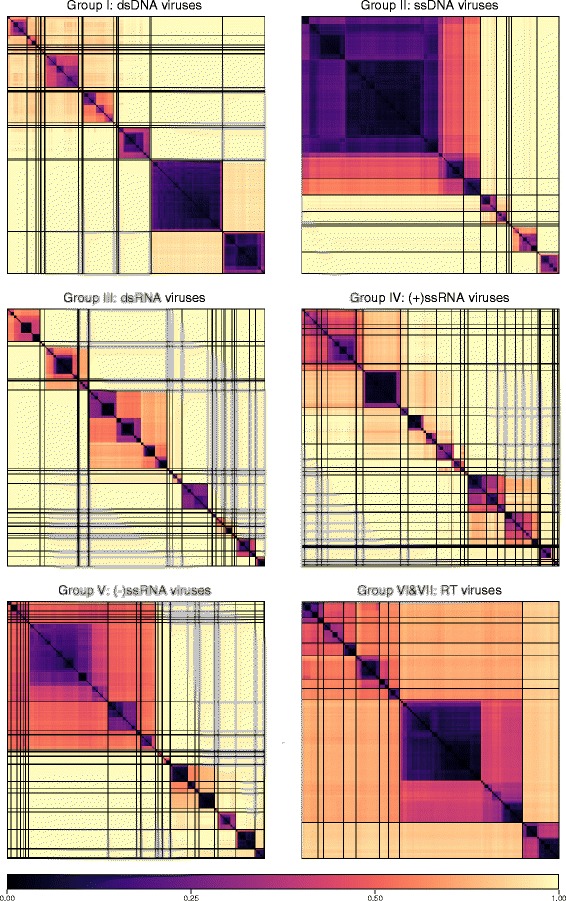
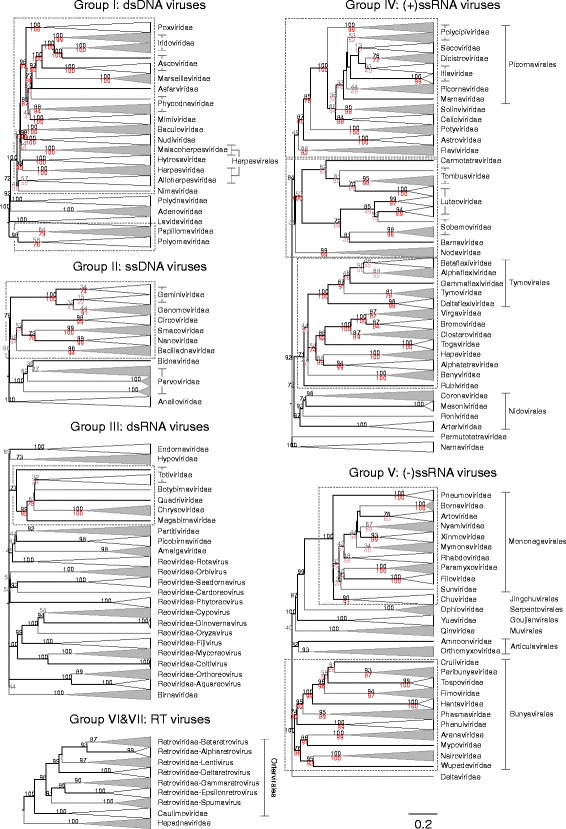
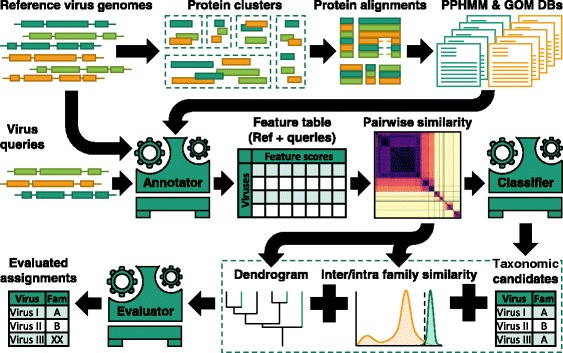
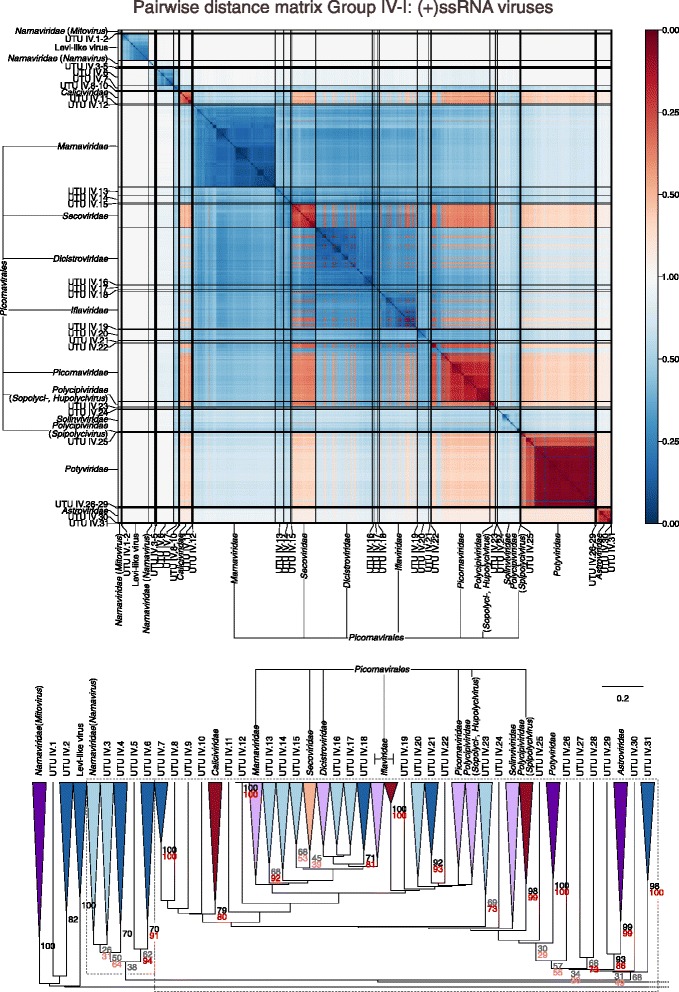
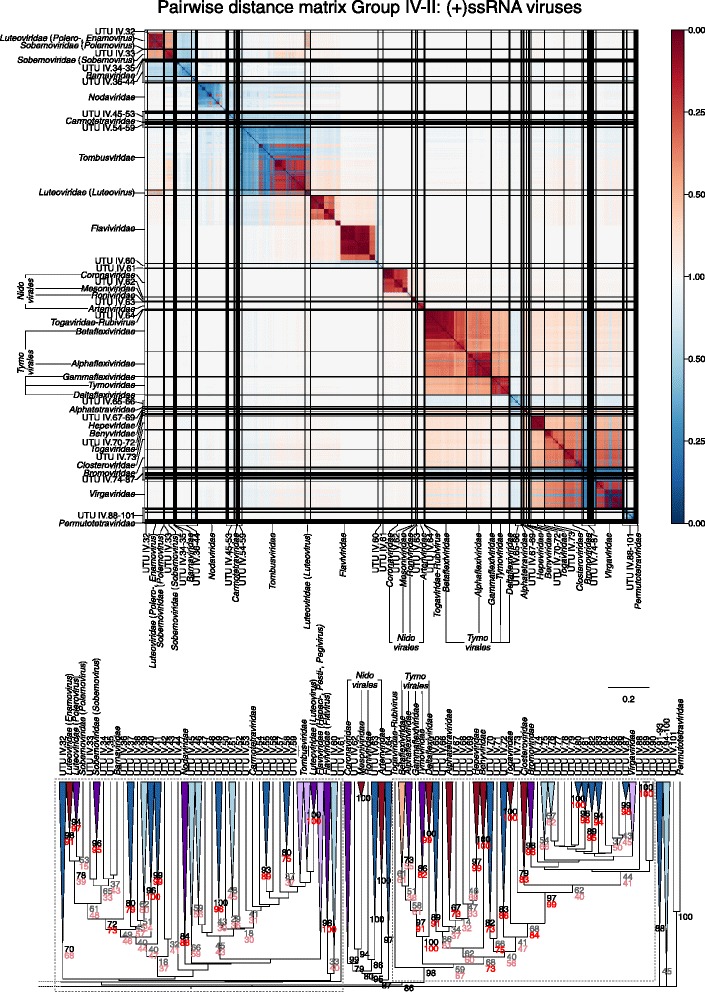
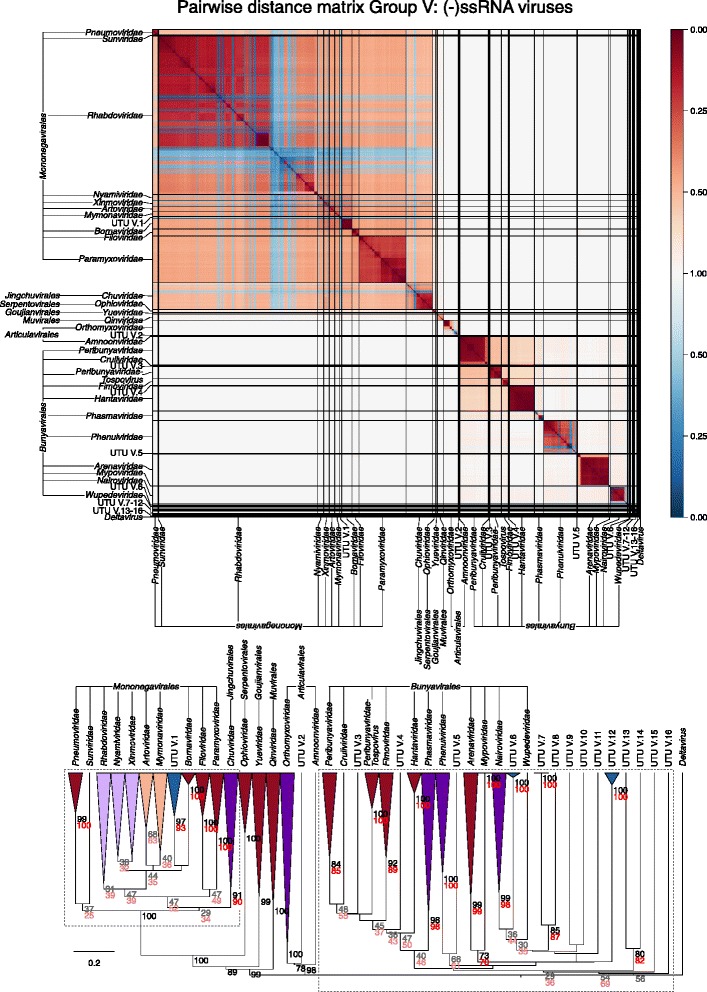
Results: The current study sought to determine whether the existing virus taxonomy could be reproduced by examination of genetic relationships through the extraction of protein-coding gene signatures and genome organisational features. We found large-scale consistency between genetic relationships and taxonomic assignments for viruses of all genome configurations and genome sizes. The analysis pipeline that we have called 'Genome Relationships Applied to Virus Taxonomy' (GRAViTy) was highly effective at reproducing the current assignments of viruses at family level as well as inter-family groupings into orders. Its ability to correctly differentiate assigned viruses from unassigned viruses, and classify them into the correct taxonomic group, was evaluated by threefold cross-validation technique. This predicted family membership of eukaryotic viruses with close to 100% accuracy and specificity potentially enabling the algorithm to predict assignments for the vast corpus of metagenomic sequences consistently with ICTV taxonomy rules. In an evaluation run of GRAViTy, over one half (460/921) of (near)-complete genome sequences from several large published metagenomic eukaryotic virus datasets were assigned to 127 novel family-level groupings. If corroborated by other analysis methods, these would potentially more than double the number of eukaryotic virus families in the ICTV taxonomy.
Conclusions: A rapid and objective means to explore metagenomic viral diversity and make informed recommendations for their assignments at each taxonomic layer is essential. GRAViTy provides one means to make rule-based assignments at family and order levels in a manner that preserves the integrity and underlying organisational principles of the current ICTV taxonomy framework. Such methods are increasingly required as the vast virosphere is explored.
Keywords: Baltimore classification; Hidden Markov model; Metagenomic; Taxon; Taxonomy; Virus; Virus classification.
Conflict of interest statement
Ethics approval
Not applicable
Consent for publication
Not applicable
Competing interests
Both authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures



References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
