

The mycotoxin phomoxanthone A disturbs the form and function of the inner mitochondrial membrane
- PMID: 29459714
- PMCID: PMC5833434
- DOI: 10.1038/s41419-018-0312-8
The mycotoxin phomoxanthone A disturbs the form and function of the inner mitochondrial membrane
Abstract
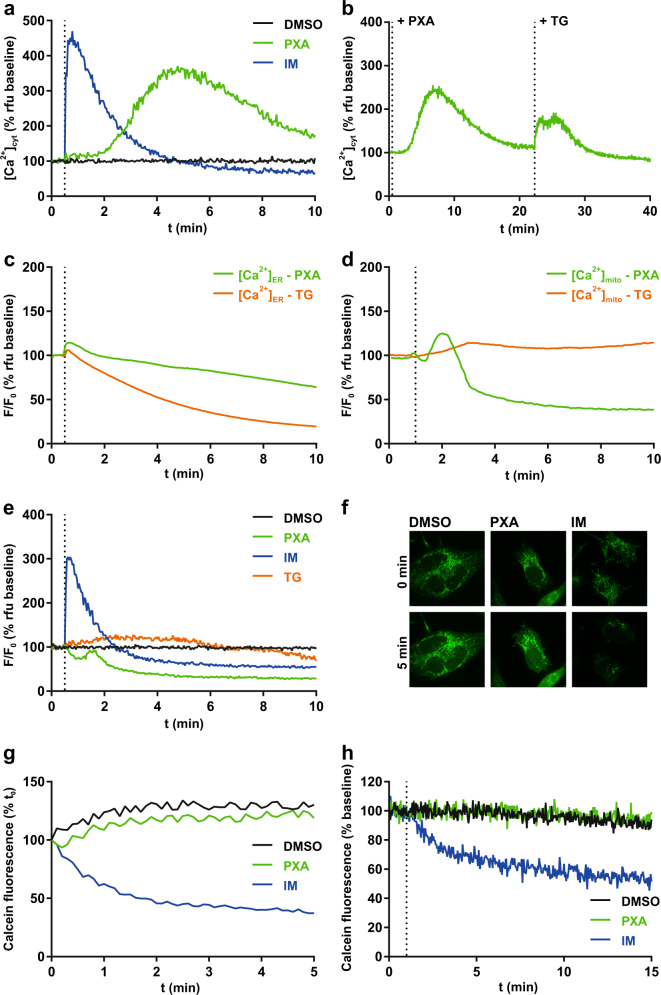
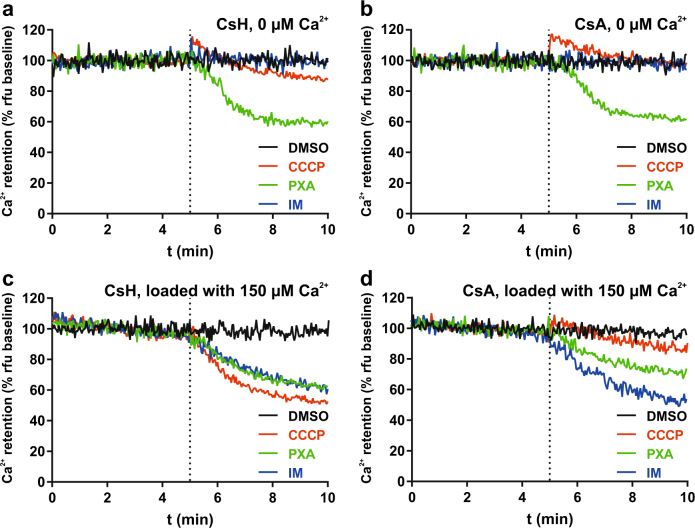
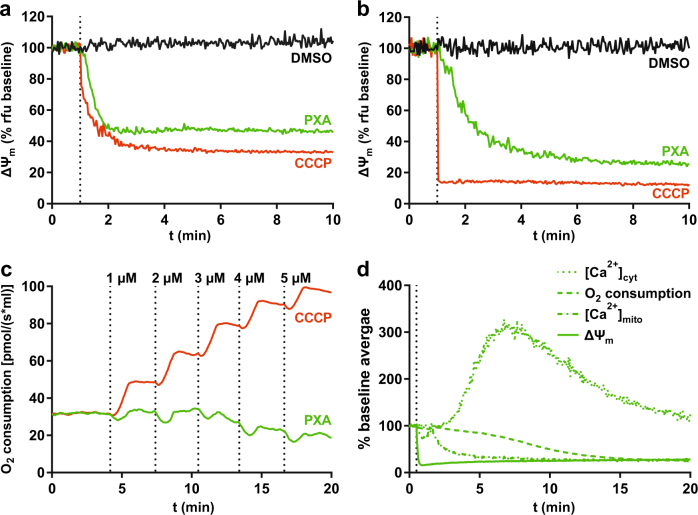
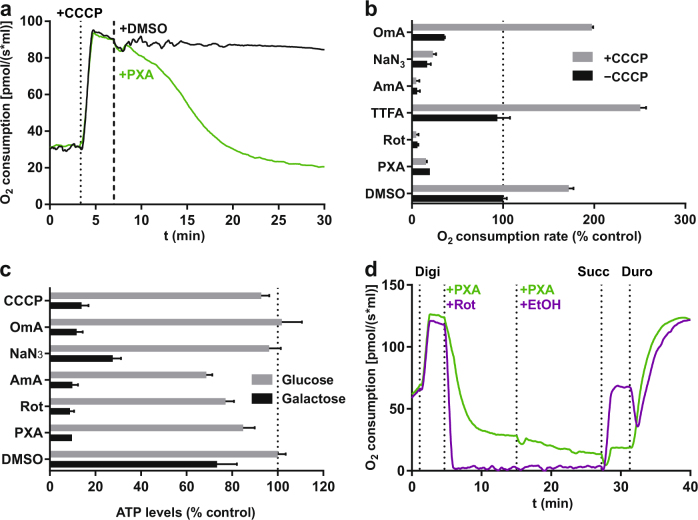
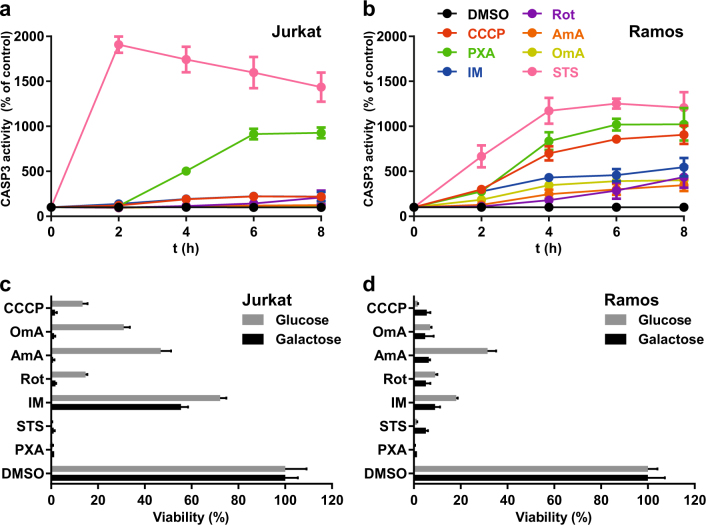
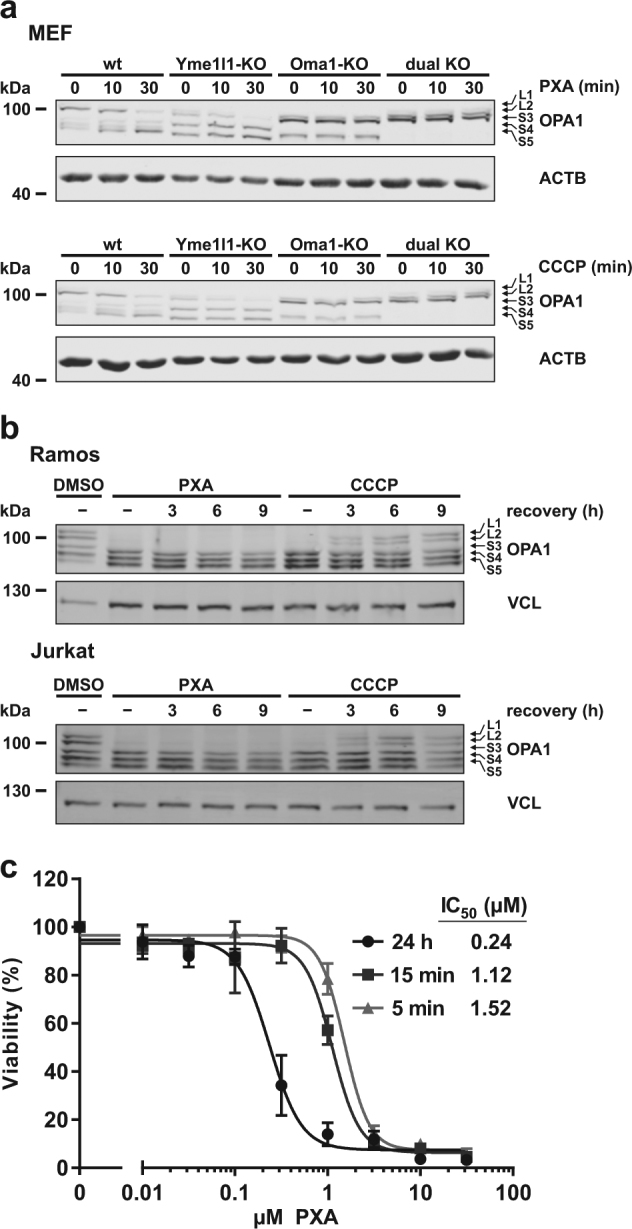
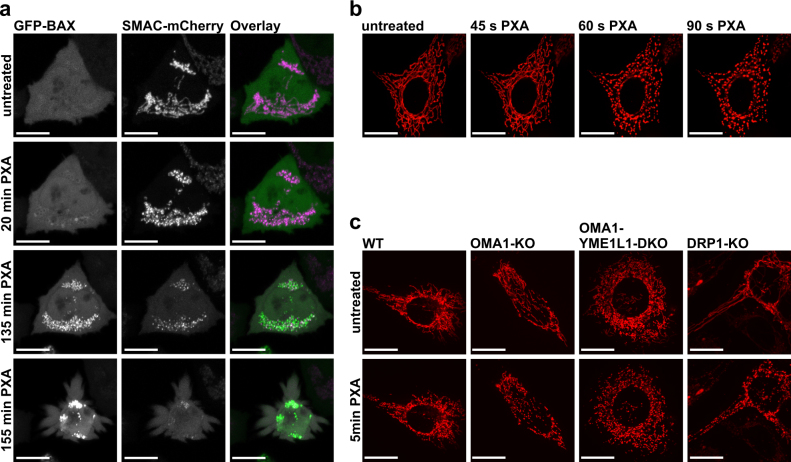
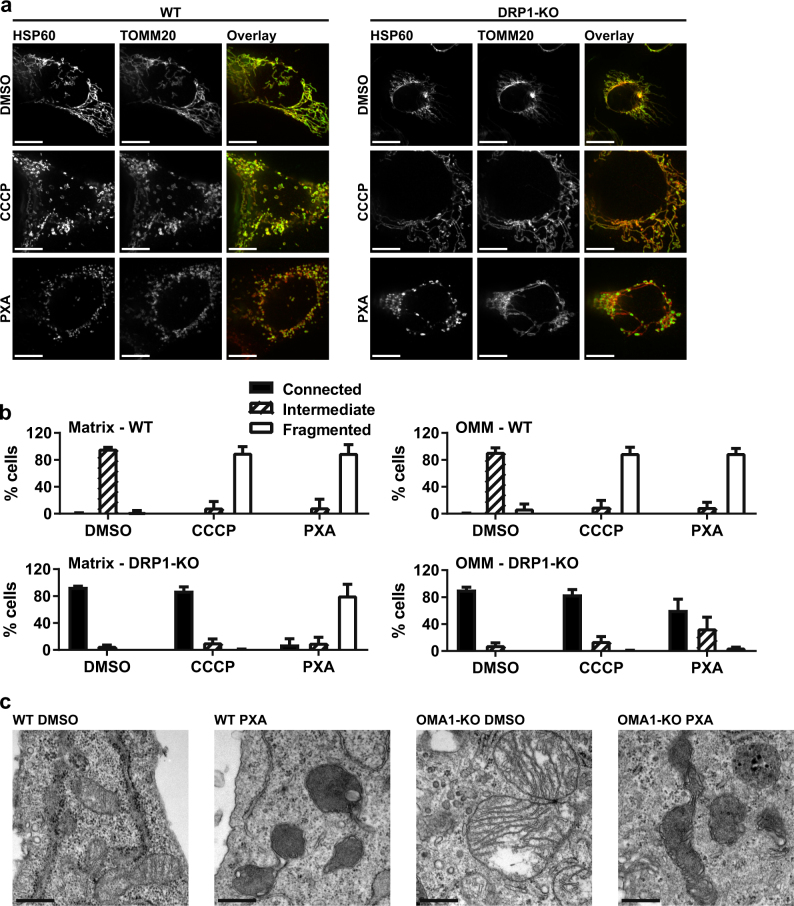
Mitochondria are cellular organelles with crucial functions in the generation and distribution of ATP, the buffering of cytosolic Ca2+ and the initiation of apoptosis. Compounds that interfere with these functions are termed mitochondrial toxins, many of which are derived from microbes, such as antimycin A, oligomycin A, and ionomycin. Here, we identify the mycotoxin phomoxanthone A (PXA), derived from the endophytic fungus Phomopsis longicolla, as a mitochondrial toxin. We show that PXA elicits a strong release of Ca2+ from the mitochondria but not from the ER. In addition, PXA depolarises the mitochondria similarly to protonophoric uncouplers such as CCCP, yet unlike these, it does not increase but rather inhibits cellular respiration and electron transport chain activity. The respiration-dependent mitochondrial network structure rapidly collapses into fragments upon PXA treatment. Surprisingly, this fragmentation is independent from the canonical mitochondrial fission and fusion mediators DRP1 and OPA1, and exclusively affects the inner mitochondrial membrane, leading to cristae disruption, release of pro-apoptotic proteins, and apoptosis. Taken together, our results suggest that PXA is a mitochondrial toxin with a novel mode of action that might prove a useful tool for the study of mitochondrial ion homoeostasis and membrane dynamics.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
