

Exploring the genetics and non-cell autonomous mechanisms underlying ALS/FTLD
- PMID: 29459769
- PMCID: PMC5864209
- DOI: 10.1038/s41418-018-0060-4
Exploring the genetics and non-cell autonomous mechanisms underlying ALS/FTLD
Abstract
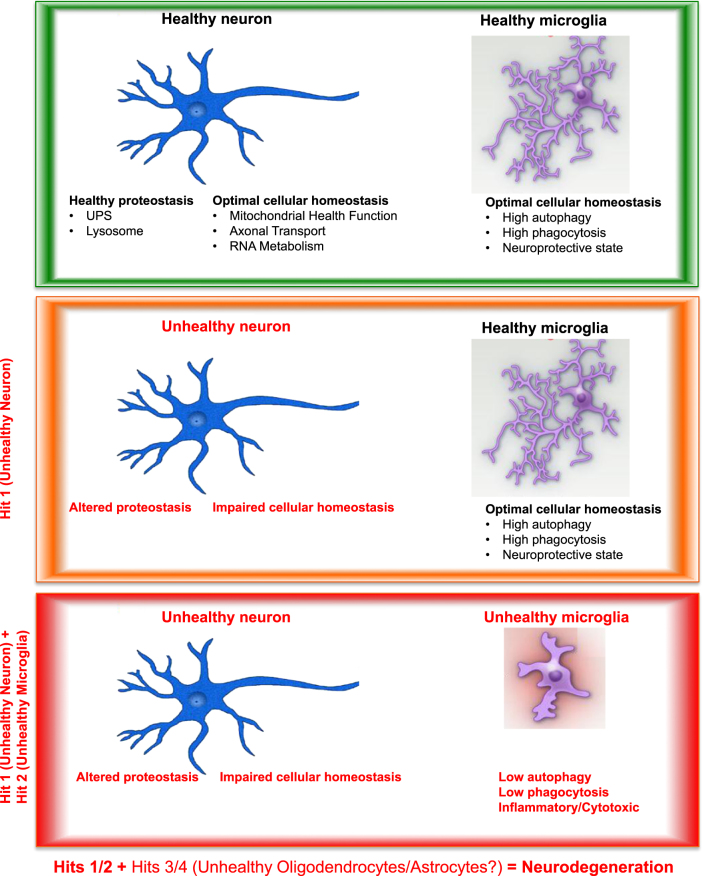
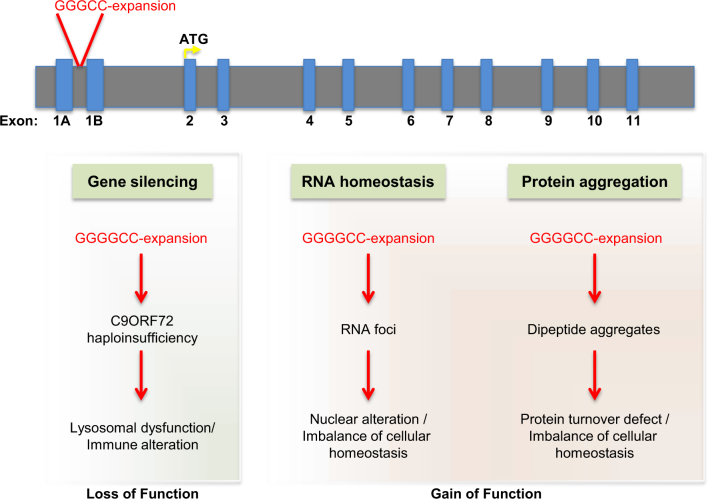
Although amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease, was first described in 1874, a flurry of genetic discoveries in the last 10 years has markedly increased our understanding of this disease. These findings have not only enhanced our knowledge of mechanisms leading to ALS, but also have revealed that ALS shares many genetic causes with another neurodegenerative disease, frontotemporal lobar dementia (FTLD). In this review, we survey how recent genetic studies have bridged our mechanistic understanding of these two related diseases and how the genetics behind ALS and FTLD point to complex disorders, implicating non-neuronal cell types in disease pathophysiology. The involvement of non-neuronal cell types is consistent with a non-cell autonomous component in these diseases. This is further supported by studies that identified a critical role of immune-associated genes within ALS/FTLD and other neurodegenerative disorders. The molecular functions of these genes support an emerging concept that various non-autonomous functions are involved in neurodegeneration. Further insights into such a mechanism(s) will ultimately lead to a better understanding of potential routes of therapeutic intervention. Facts ALS and FTLD are severe neurodegenerative disorders on the same disease spectrum. Multiple cellular processes including dysregulation of RNA homeostasis, imbalance of proteostasis, contribute to ALS/FTLD pathogenesis. Aberrant function in non-neuronal cell types, including microglia, contributes to ALS/FTLD. Strong neuroimmune and neuroinflammatory components are associated with ALS/FTLD patients. Open Questions Why can patients with similar mutations have different disease manifestations, i.e., why do C9ORF72 mutations lead to motor neuron loss in some patients while others exhibit loss of neurons in the frontotemporal lobe? Do ALS causal mutations result in microglial dysfunction and contribute to ALS/FTLD pathology? How do microglia normally act to mitigate neurodegeneration in ALS/FTLD? To what extent do cellular signaling pathways mediate non-cell autonomous communications between distinct central nervous system (CNS) cell types during disease? Is it possible to therapeutically target specific cell types in the CNS?
Conflict of interest statement
MWK, SCS are employees of Biogen. SSWH was employed and Biogen and is now an employee of GSK. DO was employed and Biogen and is now an employee of Sanofi. HC has no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
