

Formalising recall by genotype as an efficient approach to detailed phenotyping and causal inference
- PMID: 29459775
- PMCID: PMC5818506
- DOI: 10.1038/s41467-018-03109-y
Formalising recall by genotype as an efficient approach to detailed phenotyping and causal inference
Abstract
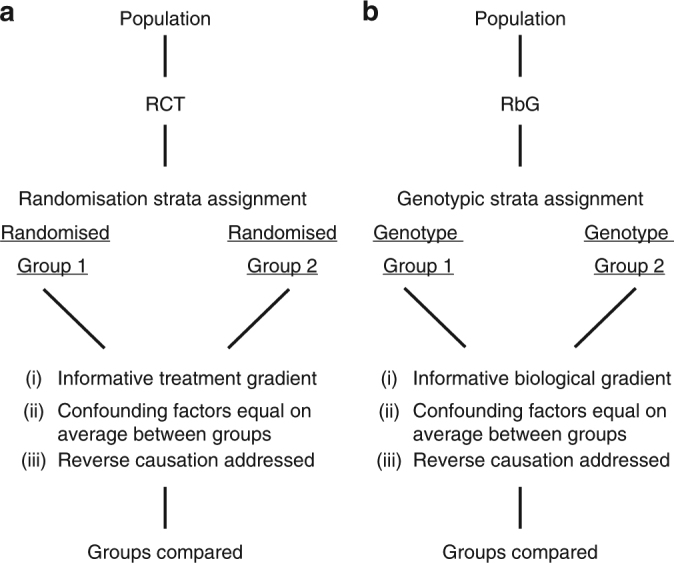
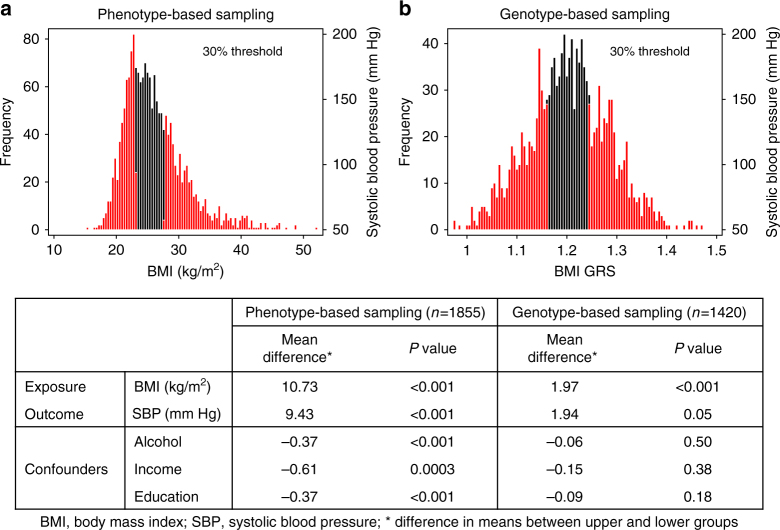
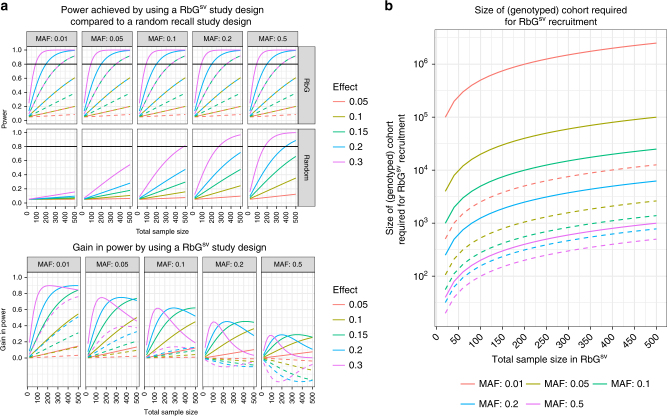
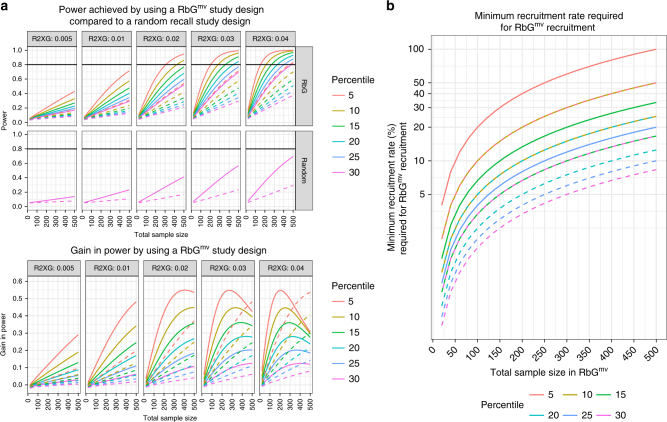
Detailed phenotyping is required to deepen our understanding of the biological mechanisms behind genetic associations. In addition, the impact of potentially modifiable risk factors on disease requires analytical frameworks that allow causal inference. Here, we discuss the characteristics of Recall-by-Genotype (RbG) as a study design aimed at addressing both these needs. We describe two broad scenarios for the application of RbG: studies using single variants and those using multiple variants. We consider the efficacy and practicality of the RbG approach, provide a catalogue of UK-based resources for such studies and present an online RbG study planner.
Conflict of interest statement
TF has consulted for Boehringer Ingelheim and Sanofi and received research funding fromGSK. The remaining authors have no conflicts of interest.
Figures
References
Publication types
MeSH terms
Grants and funding
- MC_U123292700/MRC_/Medical Research Council/United Kingdom
- G0600705/MRC_/Medical Research Council/United Kingdom
- R01 DK098032/DK/NIDDK NIH HHS/United States
- MC_UU_12012/5/MRC_/Medical Research Council/United Kingdom
- MC_EX_MR/M01424X/1/MRC_/Medical Research Council/United Kingdom
- MR/L003120/1/MRC_/Medical Research Council/United Kingdom
- U01 DK105535/DK/NIDDK NIH HHS/United States
- MC_PC_MR/R020183/1/MRC_/Medical Research Council/United Kingdom
- MC_PC_15018/MRC_/Medical Research Council/United Kingdom
- RG/16/4/32218/BHF_/British Heart Foundation/United Kingdom
- MC_UU_12013/1/MRC_/Medical Research Council/United Kingdom
- MR/P02811X/1/MRC_/Medical Research Council/United Kingdom
- G0902313/MRC_/Medical Research Council/United Kingdom
- MR/P00167X/1/MRC_/Medical Research Council/United Kingdom
- G9815508/MRC_/Medical Research Council/United Kingdom
- MC_UU_12012/1/MRC_/Medical Research Council/United Kingdom
- 210561/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- 19169/CRUK_/Cancer Research UK/United Kingdom
- MC_QA137933/MRC_/Medical Research Council/United Kingdom
- MC_UU_12013/3/MRC_/Medical Research Council/United Kingdom
- MR/P013880/1/MRC_/Medical Research Council/United Kingdom
- MR/N011317/1/MRC_/Medical Research Council/United Kingdom
- MR/L020149/1/MRC_/Medical Research Council/United Kingdom
- SP/08/007/23628/BHF_/British Heart Foundation/United Kingdom
- CH/12/2/29428/BHF_/British Heart Foundation/United Kingdom
- MC_UU_12013/4/MRC_/Medical Research Council/United Kingdom
- MC_UU_00026/3/MRC_/Medical Research Council/United Kingdom
- MR/L010305/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
