

Neuroinflammation and Central Sensitization in Chronic and Widespread Pain
- PMID: 29462012
- PMCID: PMC6051899
- DOI: 10.1097/ALN.0000000000002130
Neuroinflammation and Central Sensitization in Chronic and Widespread Pain
Abstract
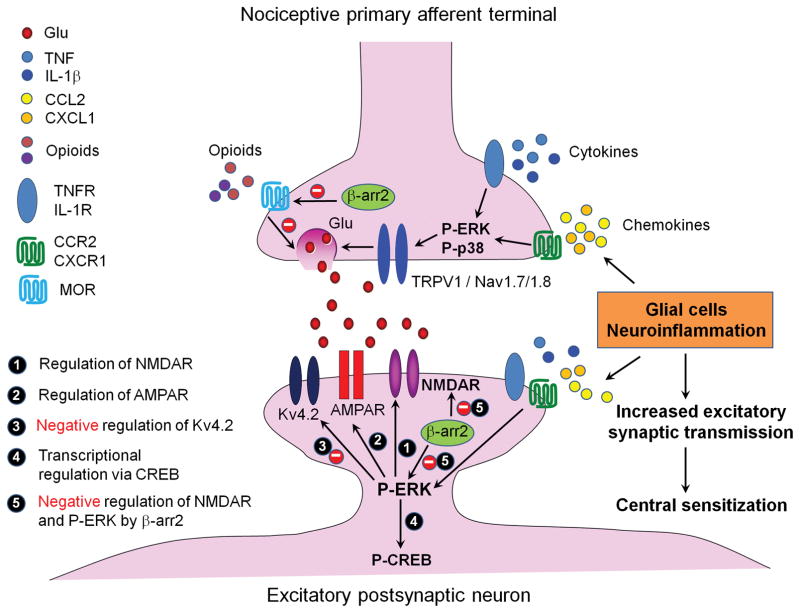
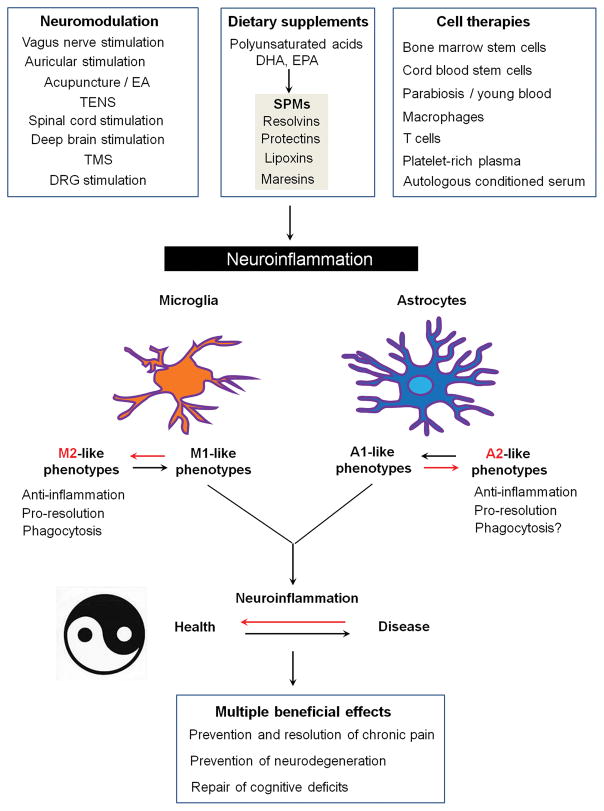
Chronic pain is maintained in part by central sensitization, a phenomenon of synaptic plasticity, and increased neuronal responsiveness in central pain pathways after painful insults. Accumulating evidence suggests that central sensitization is also driven by neuroinflammation in the peripheral and central nervous system. A characteristic feature of neuroinflammation is the activation of glial cells, such as microglia and astrocytes, in the spinal cord and brain, leading to the release of proinflammatory cytokines and chemokines. Recent studies suggest that central cytokines and chemokines are powerful neuromodulators and play a sufficient role in inducing hyperalgesia and allodynia after central nervous system administration. Sustained increase of cytokines and chemokines in the central nervous system also promotes chronic widespread pain that affects multiple body sites. Thus, neuroinflammation drives widespread chronic pain via central sensitization. We also discuss sex-dependent glial/immune signaling in chronic pain and new therapeutic approaches that control neuroinflammation for the resolution of chronic pain.
Conflict of interest statement
Figures
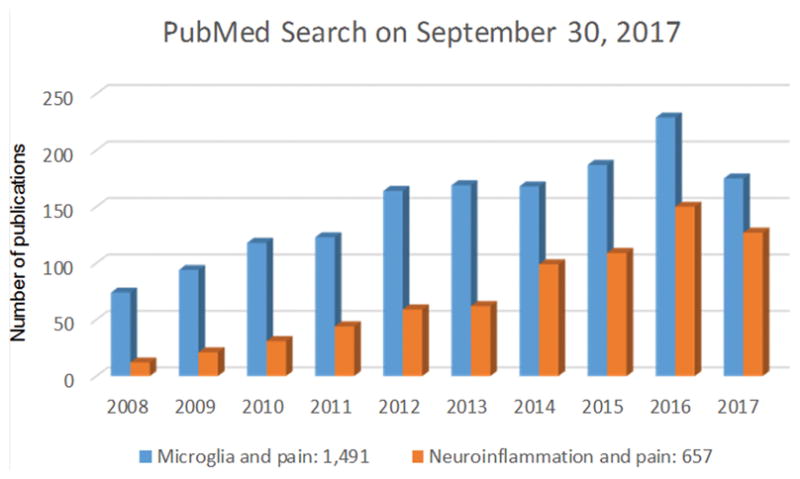
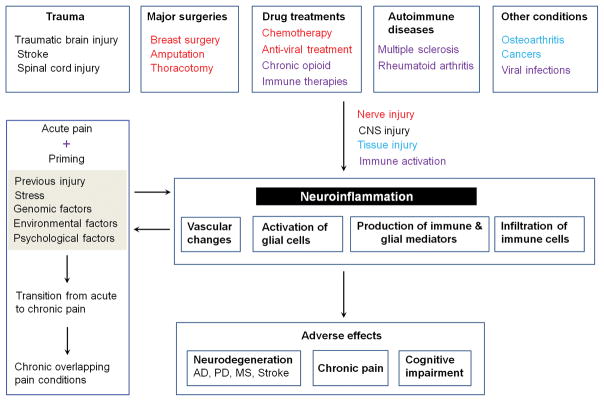
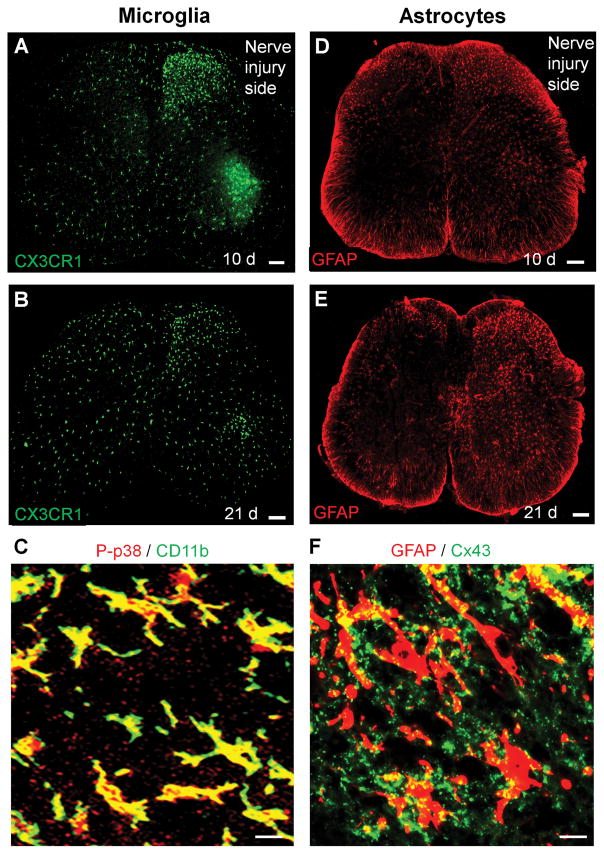
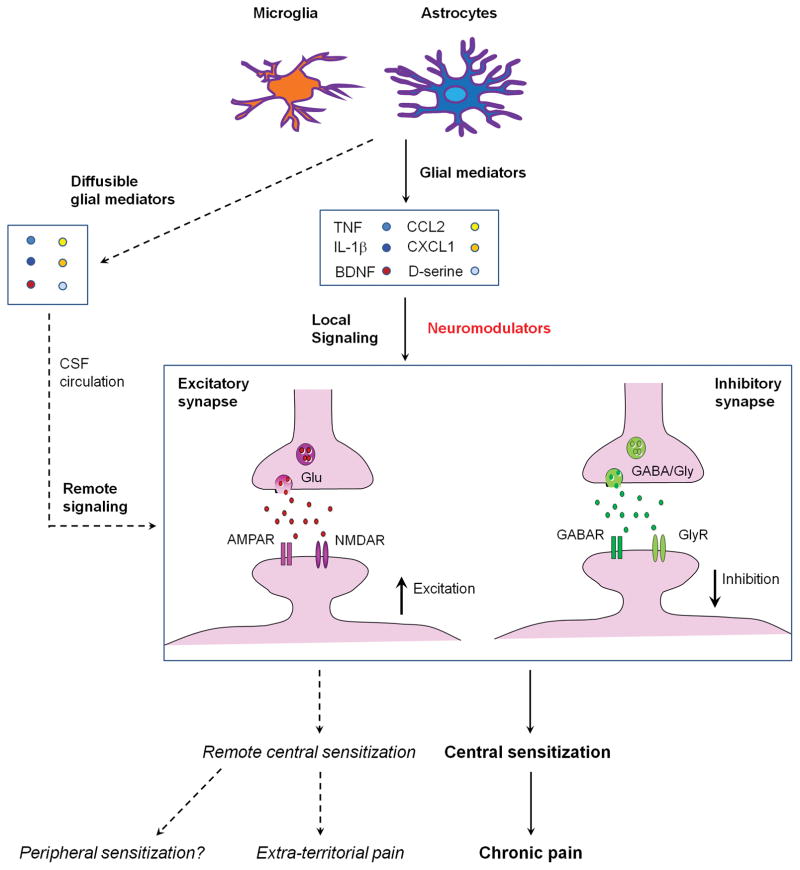
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
