

Fundamental limits on dynamic inference from single-cell snapshots
- PMID: 29463712
- PMCID: PMC5878004
- DOI: 10.1073/pnas.1714723115
Fundamental limits on dynamic inference from single-cell snapshots
Abstract
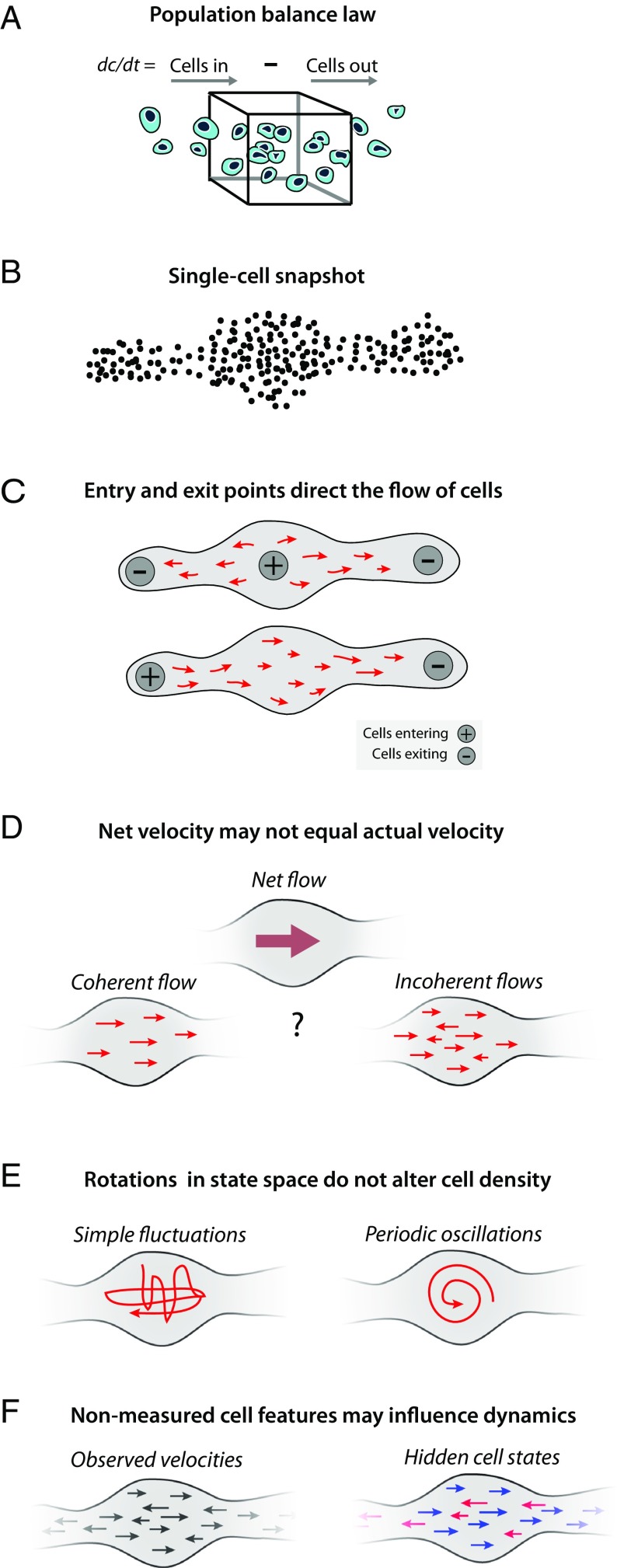
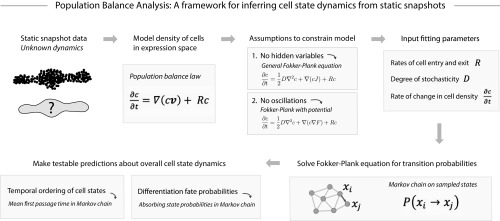
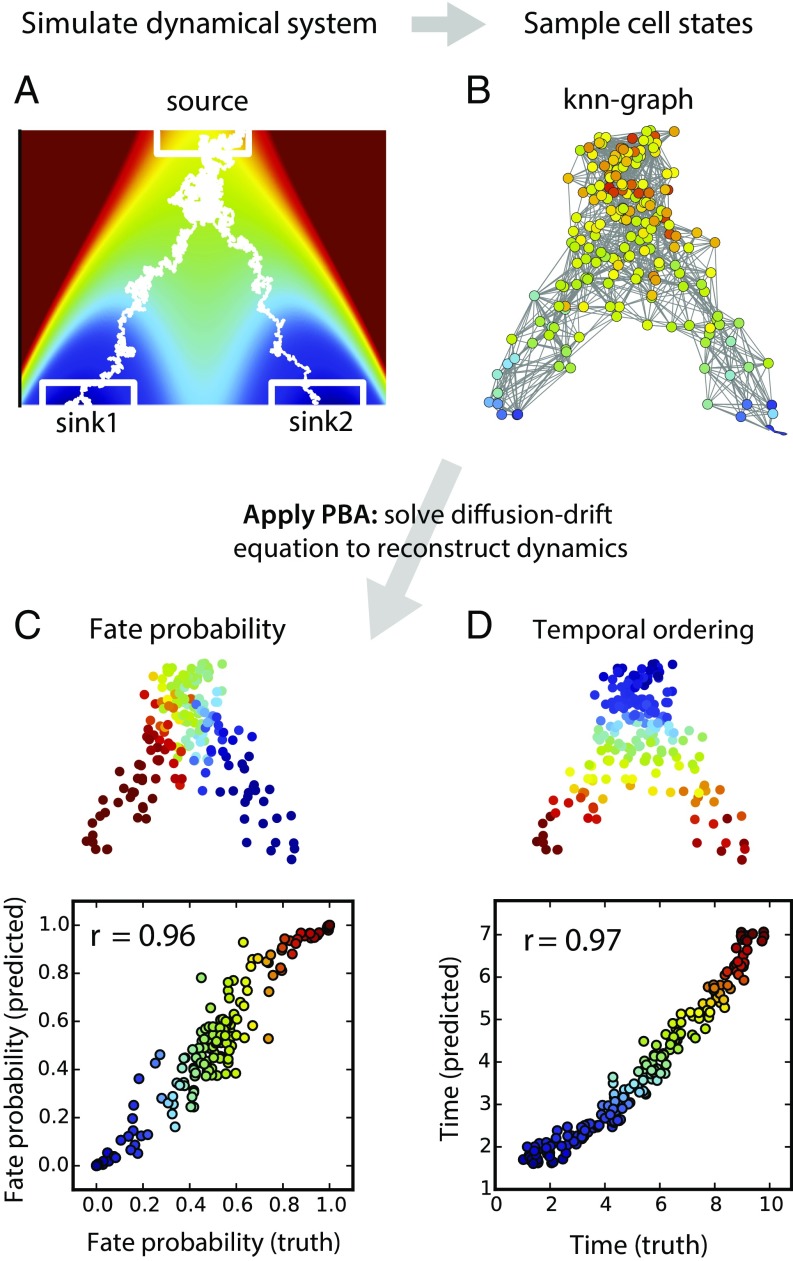
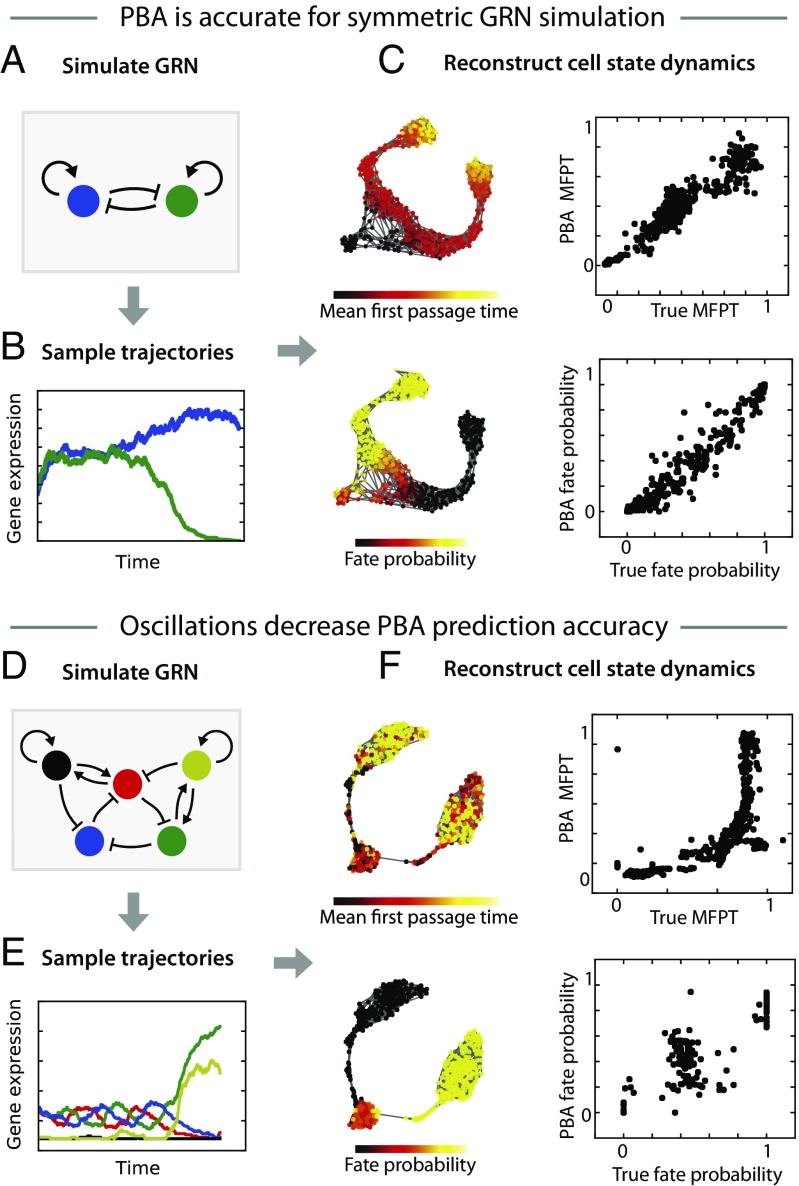
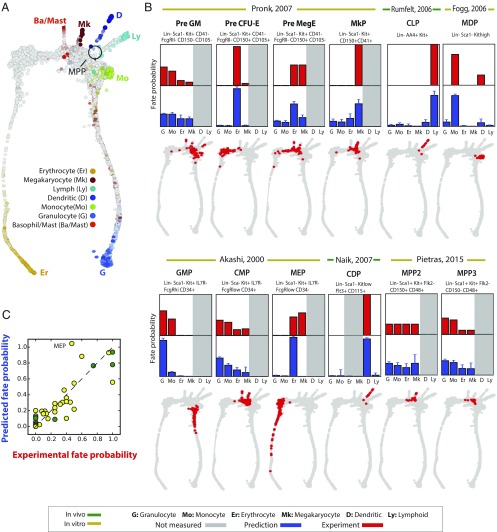
Single-cell expression profiling reveals the molecular states of individual cells with unprecedented detail. Because these methods destroy cells in the process of analysis, they cannot measure how gene expression changes over time. However, some information on dynamics is present in the data: the continuum of molecular states in the population can reflect the trajectory of a typical cell. Many methods for extracting single-cell dynamics from population data have been proposed. However, all such attempts face a common limitation: for any measured distribution of cell states, there are multiple dynamics that could give rise to it, and by extension, multiple possibilities for underlying mechanisms of gene regulation. Here, we describe the aspects of gene expression dynamics that cannot be inferred from a static snapshot alone and identify assumptions necessary to constrain a unique solution for cell dynamics from static snapshots. We translate these constraints into a practical algorithmic approach, population balance analysis (PBA), which makes use of a method from spectral graph theory to solve a class of high-dimensional differential equations. We use simulations to show the strengths and limitations of PBA, and then apply it to single-cell profiles of hematopoietic progenitor cells (HPCs). Cell state predictions from this analysis agree with HPC fate assays reported in several papers over the past two decades. By highlighting the fundamental limits on dynamic inference faced by any method, our framework provides a rigorous basis for dynamic interpretation of a gene expression continuum and clarifies best experimental designs for trajectory reconstruction from static snapshot measurements.
Keywords: dynamic inference; hematopoiesis; pseudotime; single cell; spectral graph theory.
Copyright © 2018 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
