

Population snapshots predict early haematopoietic and erythroid hierarchies
- PMID: 29466336
- PMCID: PMC5899604
- DOI: 10.1038/nature25741
Population snapshots predict early haematopoietic and erythroid hierarchies
Abstract
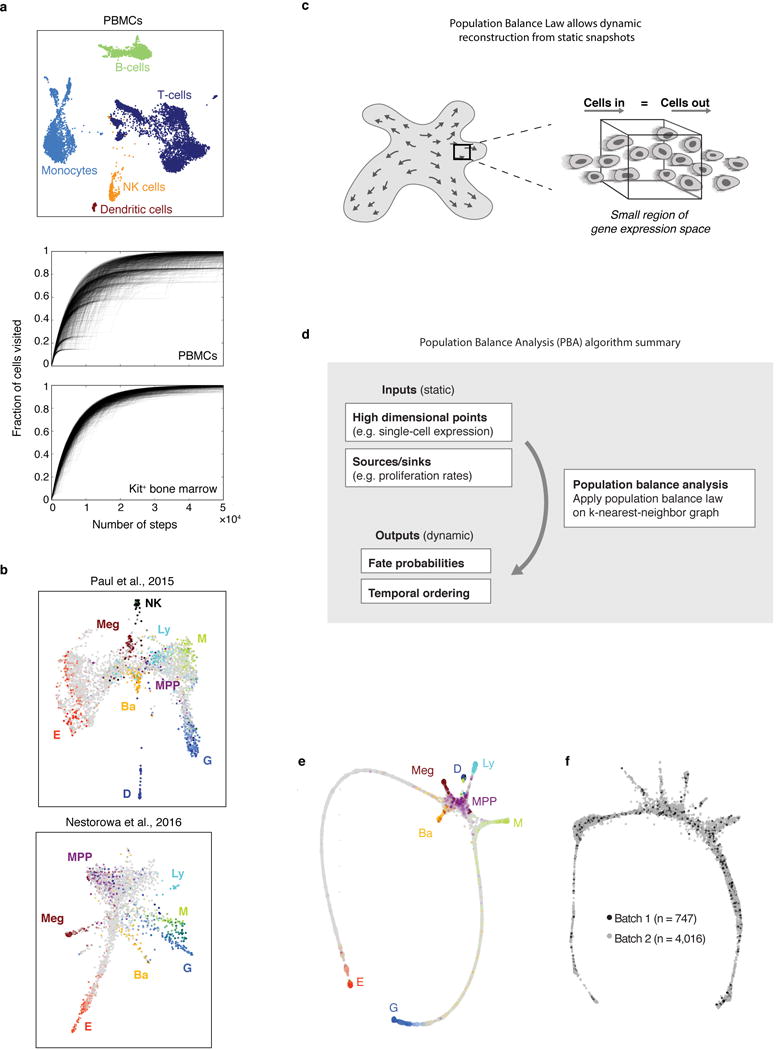
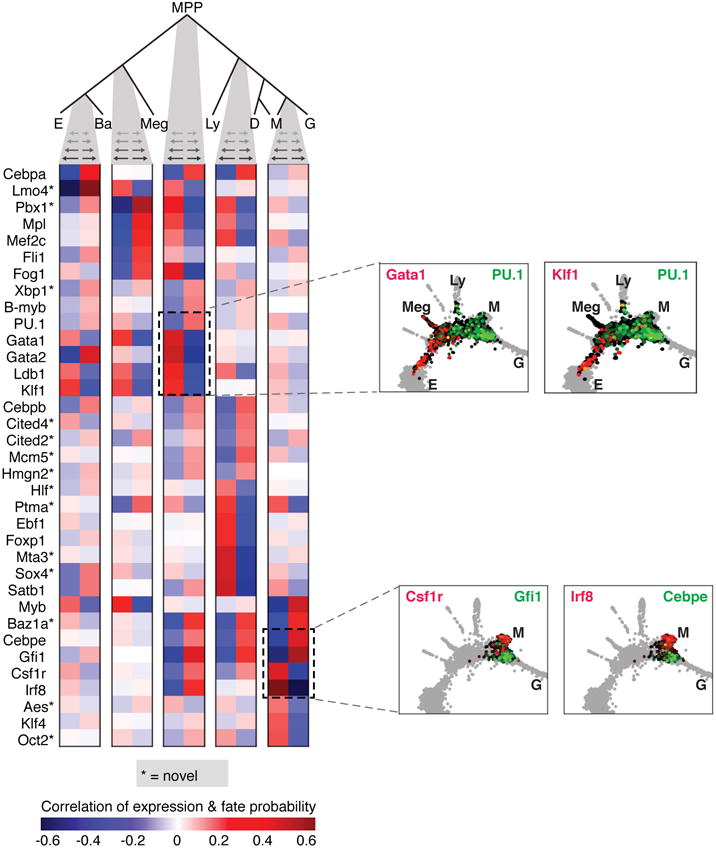
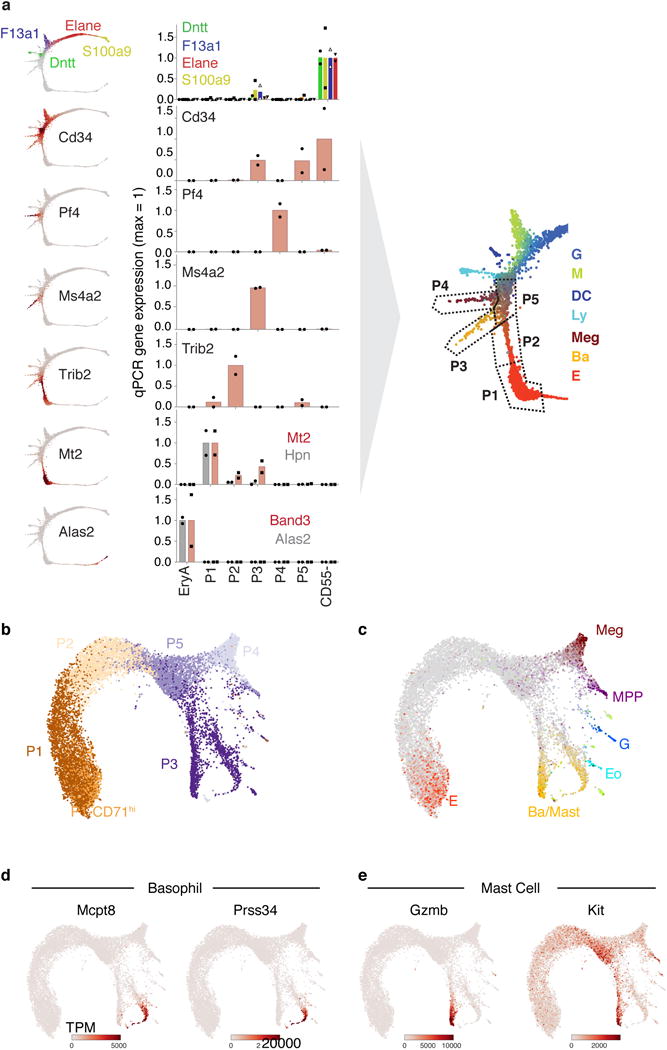
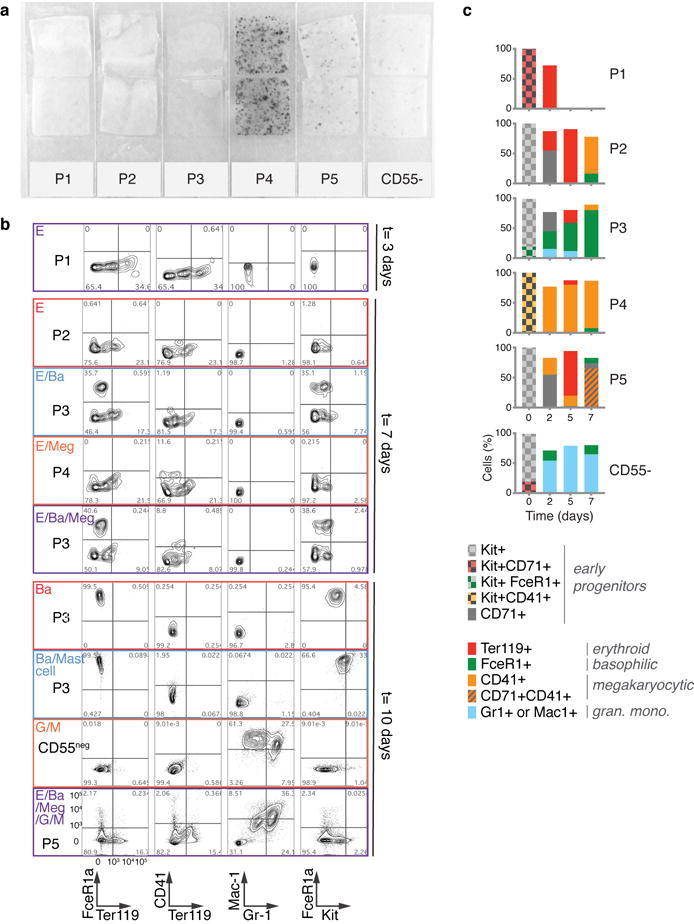
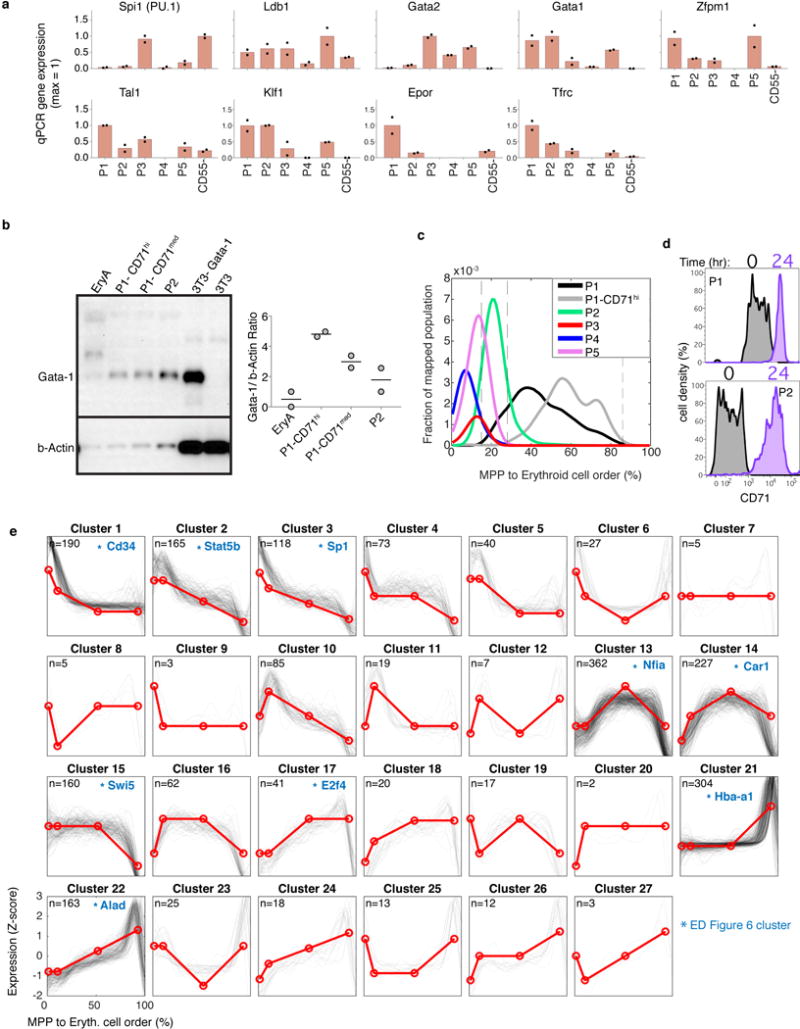
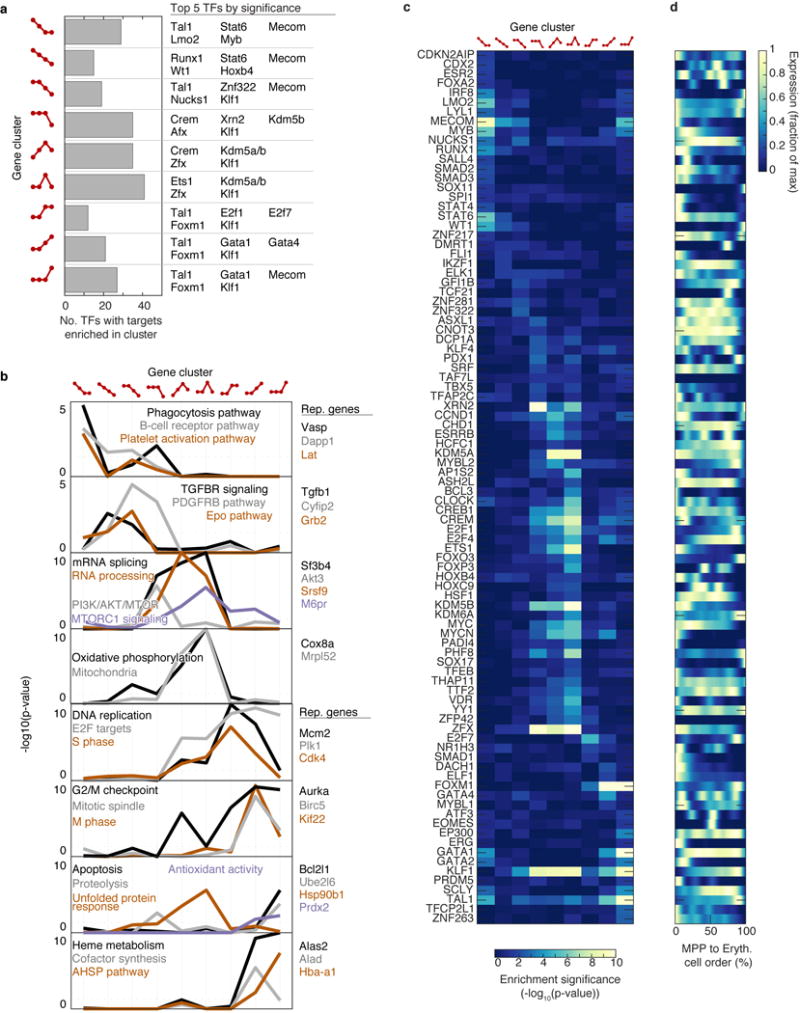
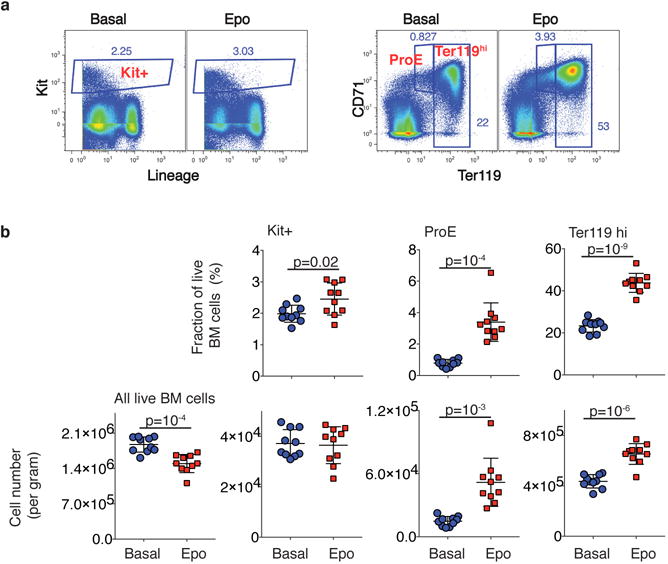
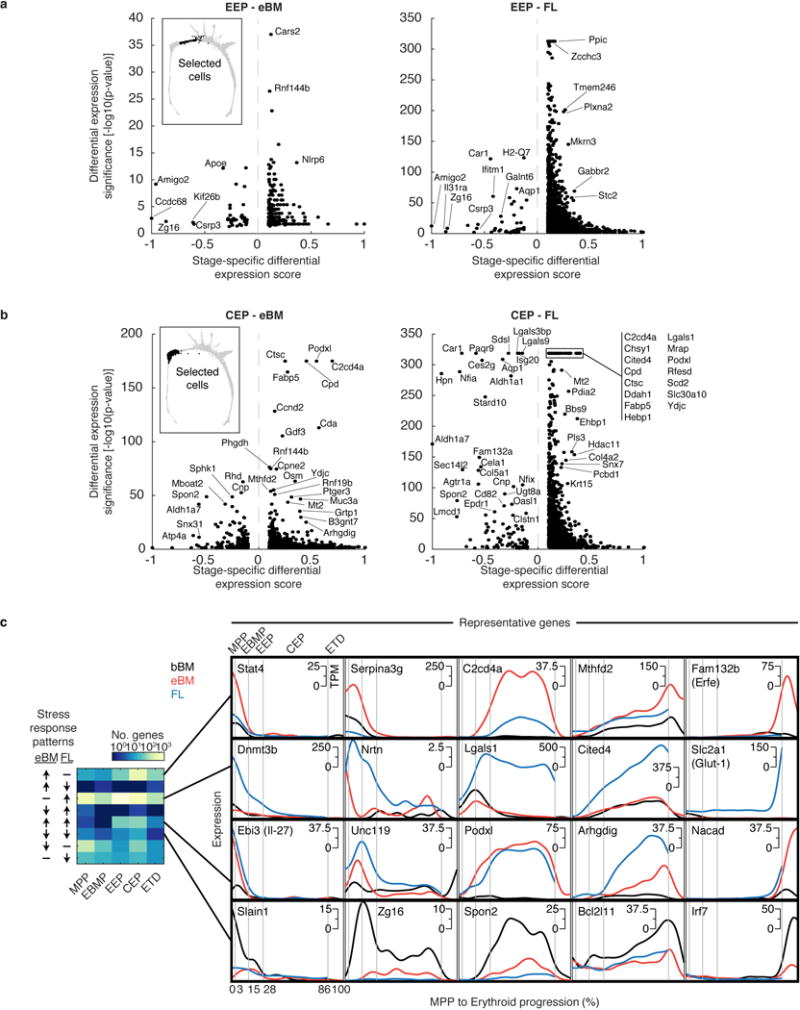
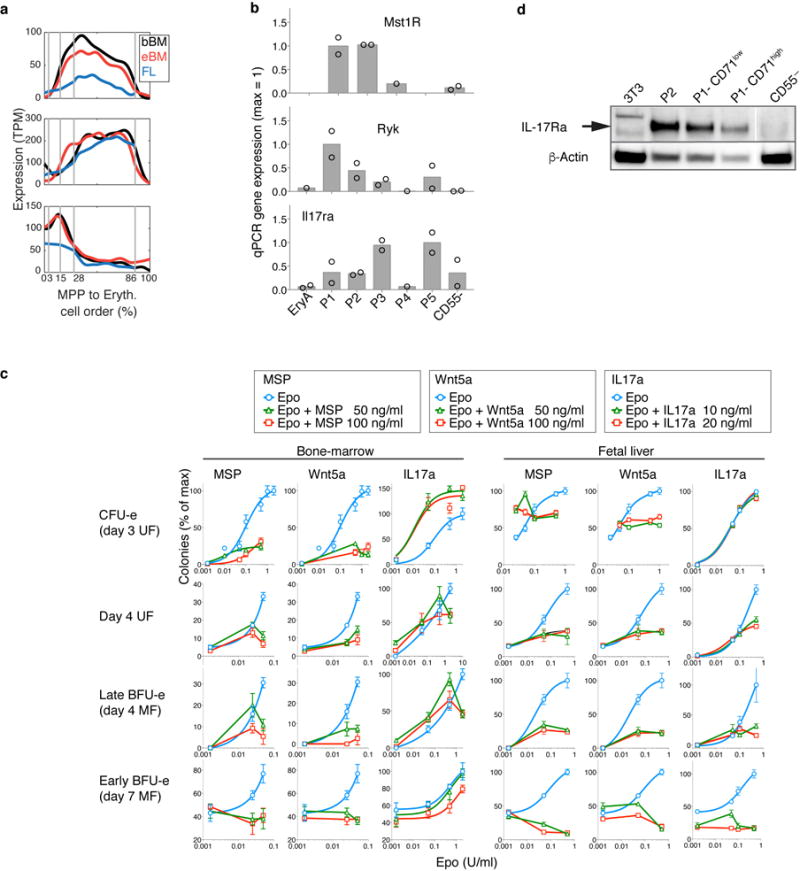
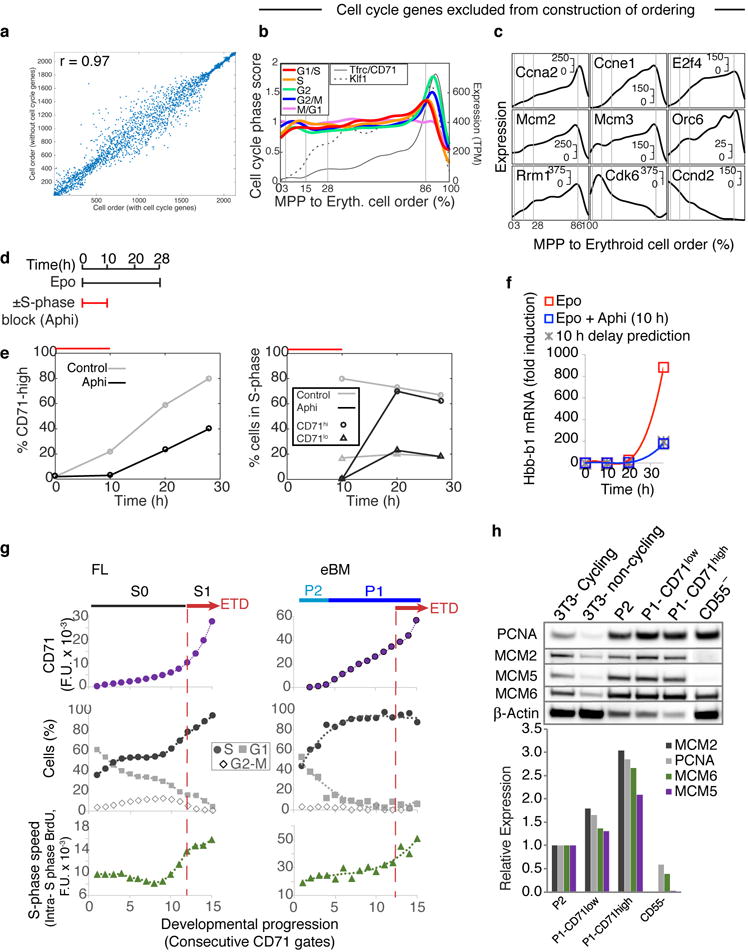
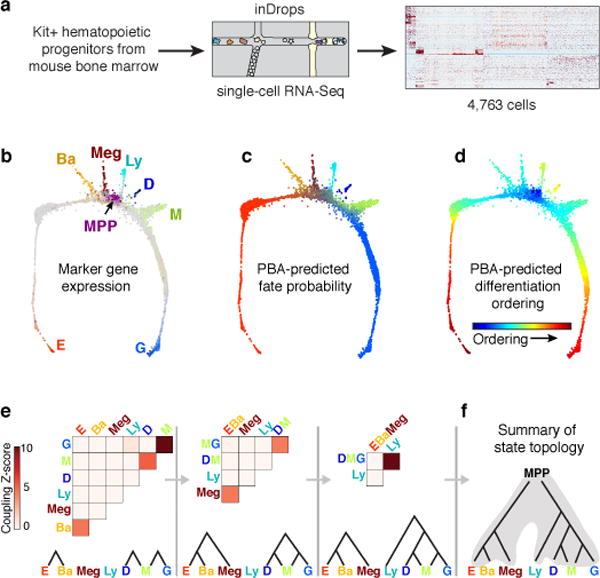
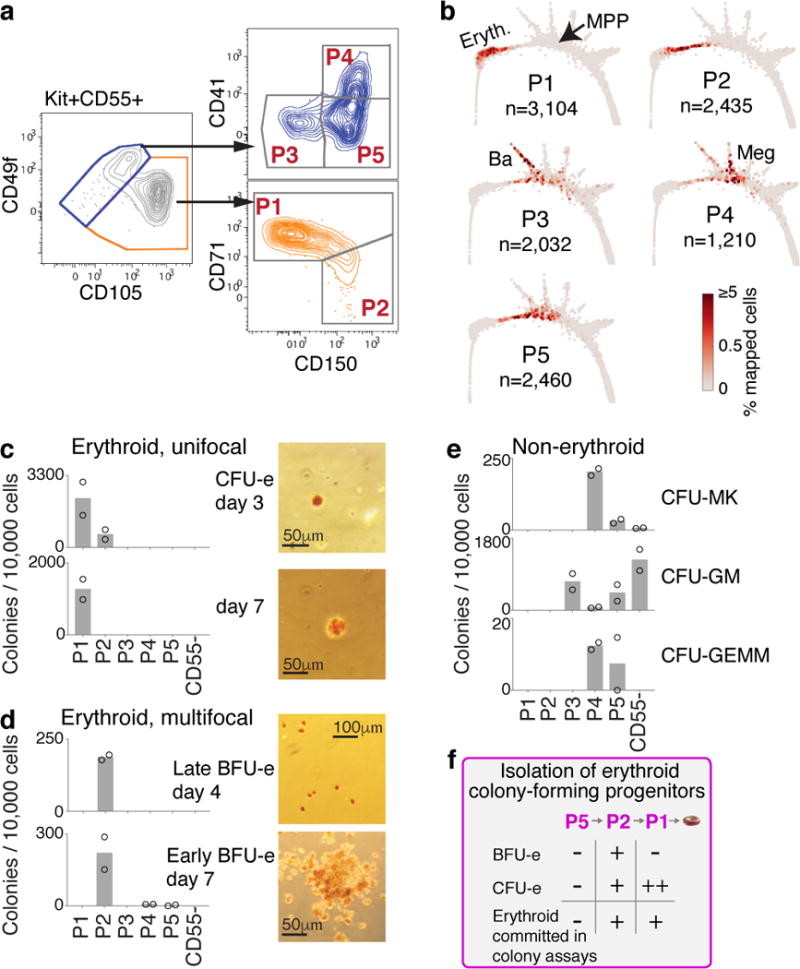
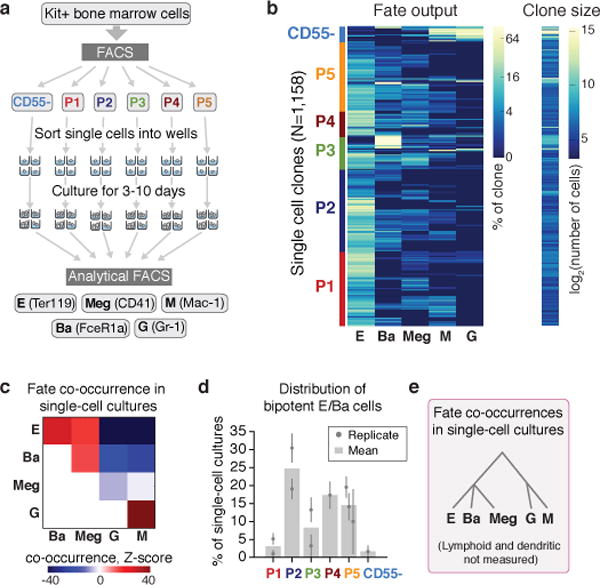
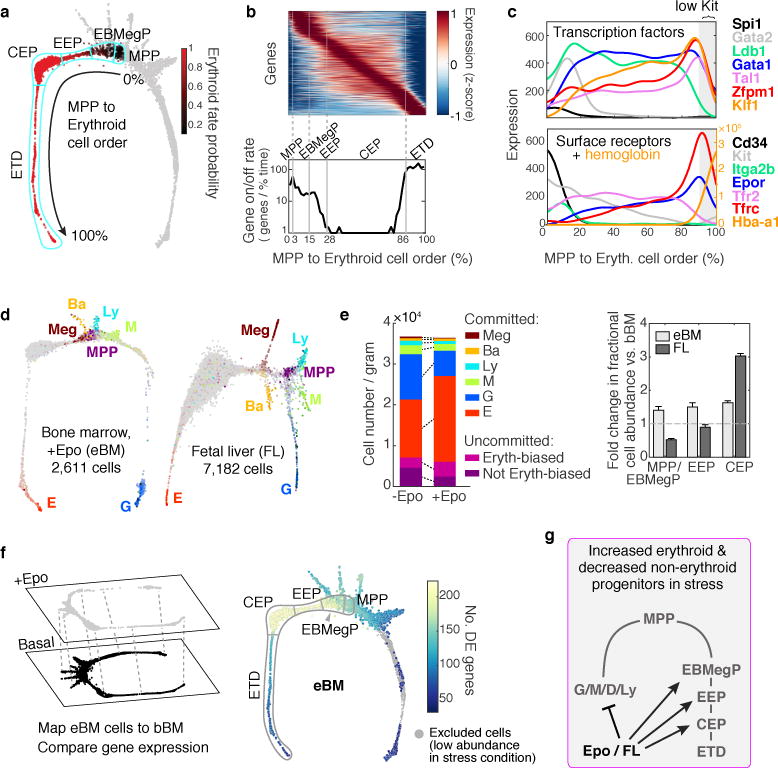
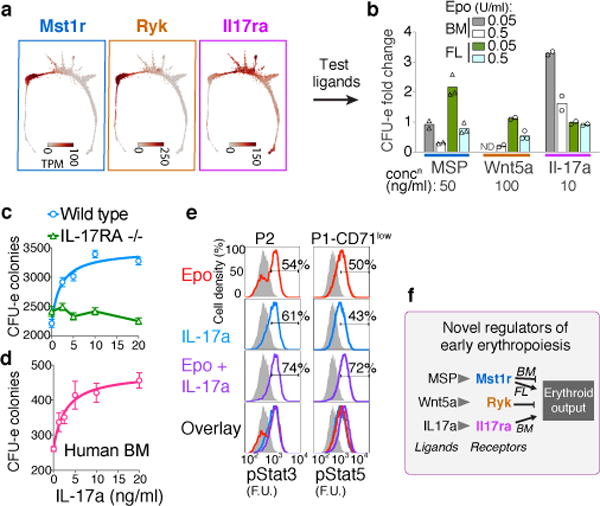
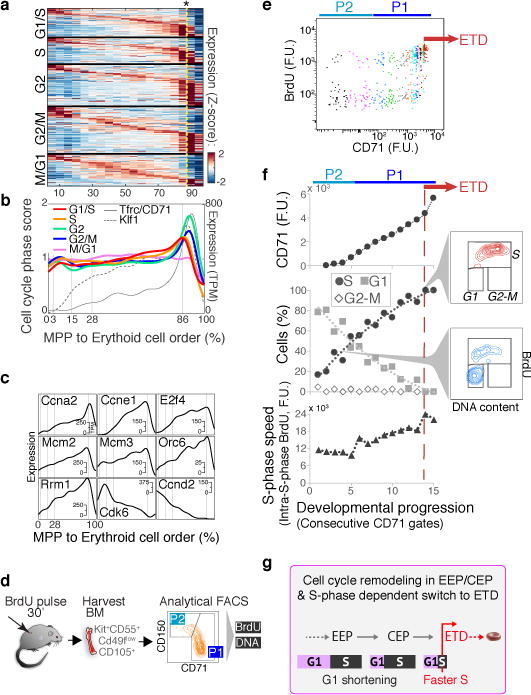
The formation of red blood cells begins with the differentiation of multipotent haematopoietic progenitors. Reconstructing the steps of this differentiation represents a general challenge in stem-cell biology. Here we used single-cell transcriptomics, fate assays and a theory that allows the prediction of cell fates from population snapshots to demonstrate that mouse haematopoietic progenitors differentiate through a continuous, hierarchical structure into seven blood lineages. We uncovered coupling between the erythroid and the basophil or mast cell fates, a global haematopoietic response to erythroid stress and novel growth factor receptors that regulate erythropoiesis. We defined a flow cytometry sorting strategy to purify early stages of erythroid differentiation, completely isolating classically defined burst-forming and colony-forming progenitors. We also found that the cell cycle is progressively remodelled during erythroid development and during a sharp transcriptional switch that ends the colony-forming progenitor stage and activates terminal differentiation. Our work showcases the utility of linking transcriptomic data to predictive fate models, and provides insights into lineage development in vivo.
Conflict of interest statement
Competing Interests: AMK is a co-founder of 1Cell-Bio.
Figures
References
-
- Hara H, Ogawa M. Erythropoietic precursors in mice under erythropoietic stimulation and suppression. Exp Hematol. 1977;5:141–148. - PubMed
-
- Gregory CJ, McCulloch EA, Till JE. The cellular basis for the defect in haemopoiesis in flexed-tailed mice. III. Restriction of the defect to erythropoietic progenitors capable of transient colony formation in vivo. Br J Haematol. 1975;30:401–410. - PubMed
Additional References for Methods section
-
- Zilionis R, et al. Single-cell barcoding and sequencing using droplet microfluidics. Nat Protocols. 2017;12:44–73. doi: 10.1038/nprot.2016.154. http://www.nature.com/nprot/journal/v12/n1/abs/nprot.2016.154.html-suppl.... - DOI - PubMed
-
- Ester M, Kriegel H, Sander J, Xu XI. Conf on Knowledge Discovery and Data Mining. :226.
-
- Daszykowski M, Walczak B, Massart DL. Looking for natural patterns in data: Part 1. Density-based approach. Chemometrics and Intelligent Laboratory Systems. 2001;56:83–92. doi: http://dx.doi.org/10.1016/S0169-7439(01)00111-3. - DOI
-
- van der Maaten L. Accelerating t-SNE using Tree-Based Algorithms. Journal of Machine Learning Research. 2014;15:3221–3245.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
