

Stroke-Like Episodes and Cerebellar Syndrome in Phosphomannomutase Deficiency (PMM2-CDG): Evidence for Hypoglycosylation-Driven Channelopathy
- PMID: 29470411
- PMCID: PMC5855841
- DOI: 10.3390/ijms19020619
Stroke-Like Episodes and Cerebellar Syndrome in Phosphomannomutase Deficiency (PMM2-CDG): Evidence for Hypoglycosylation-Driven Channelopathy
Abstract
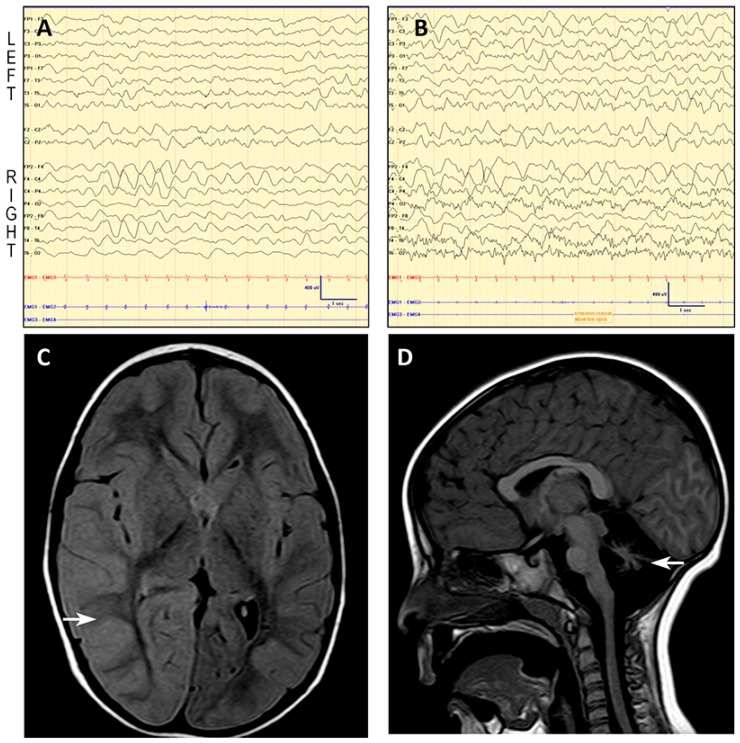
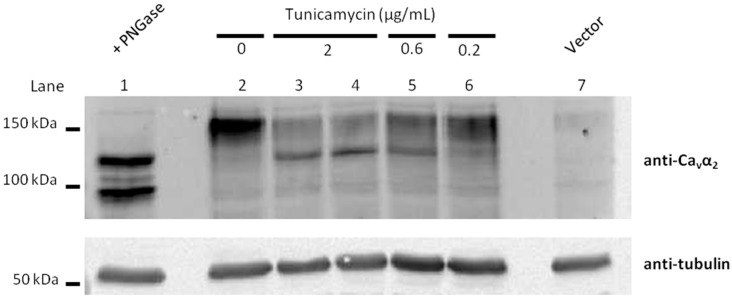
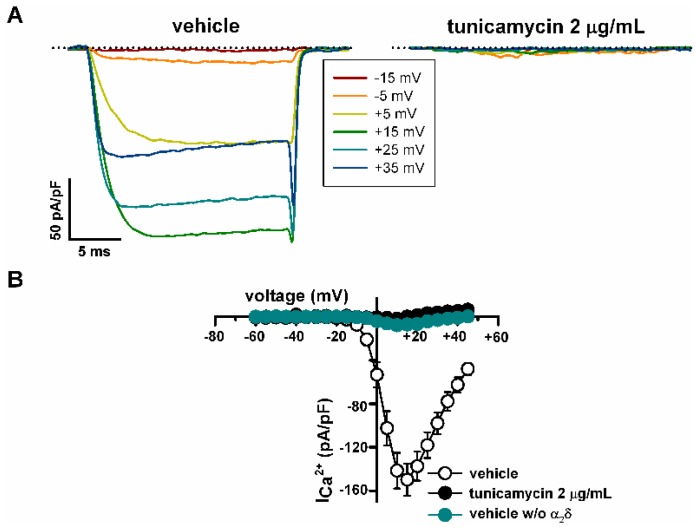
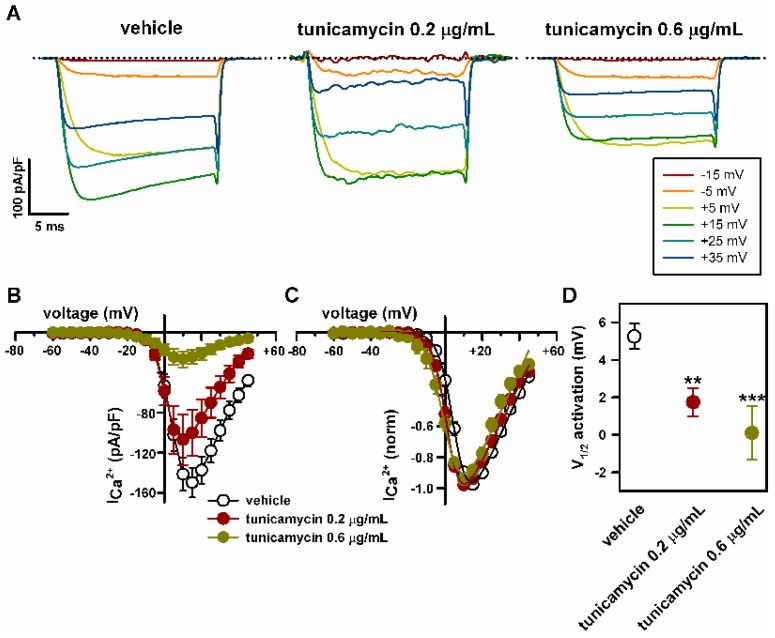
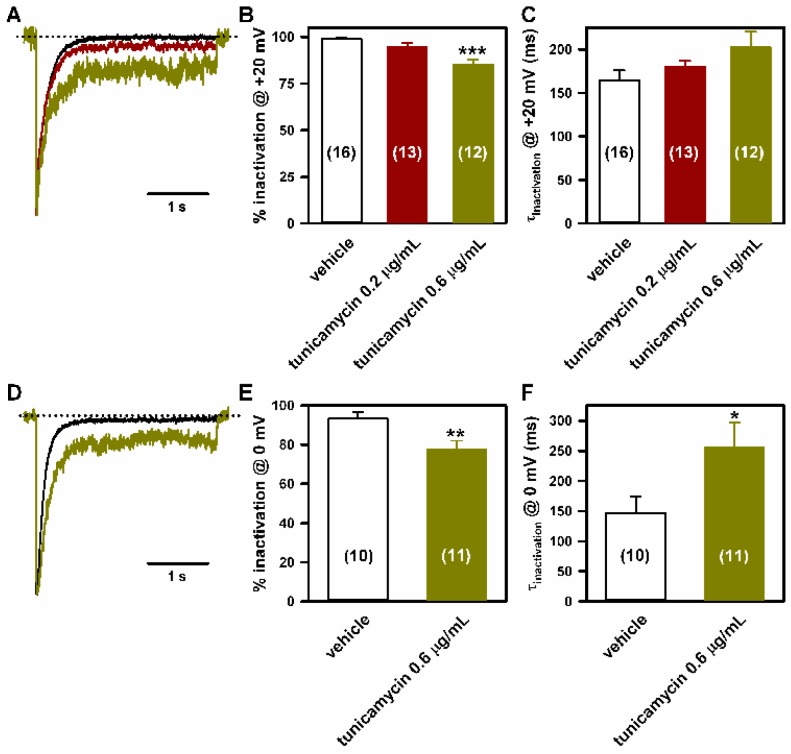
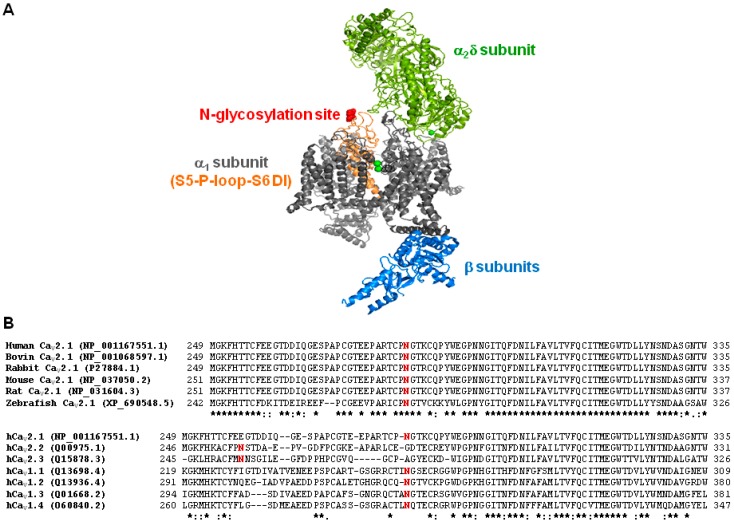
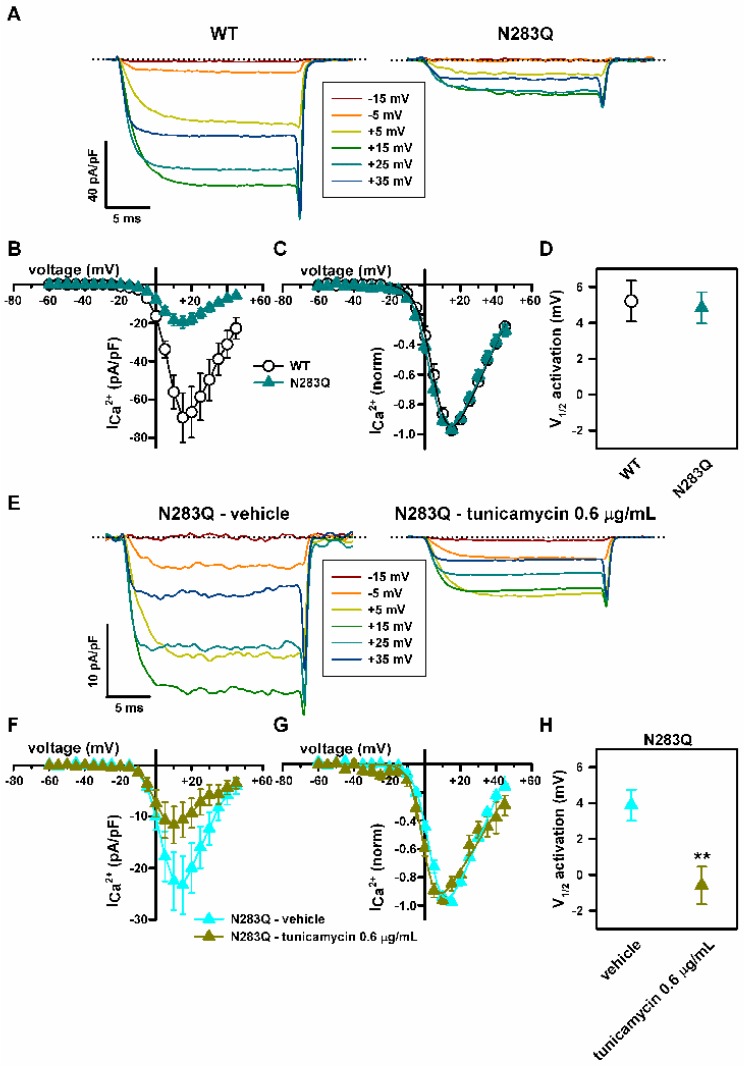
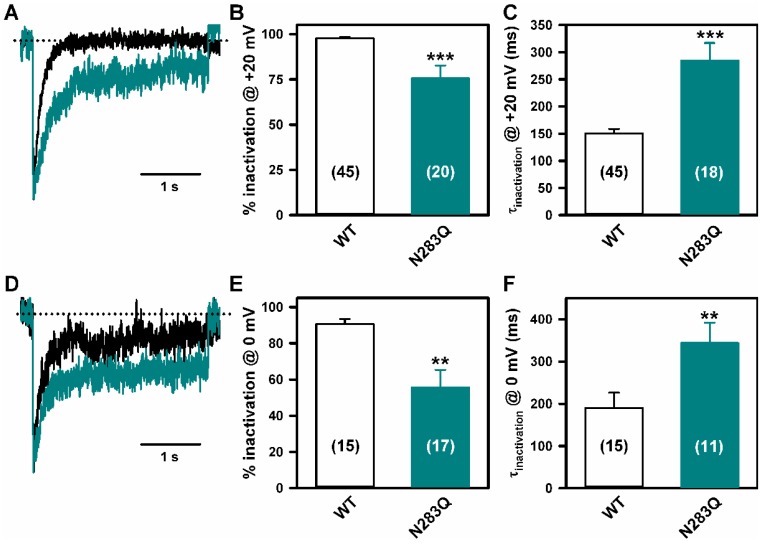
Stroke-like episodes (SLE) occur in phosphomannomutase deficiency (PMM2-CDG), and may complicate the course of channelopathies related to Familial Hemiplegic Migraine (FHM) caused by mutations in CACNA1A (encoding CaV2.1 channel). The underlying pathomechanisms are unknown. We analyze clinical variables to detect risk factors for SLE in a series of 43 PMM2-CDG patients. We explore the hypothesis of abnormal CaV2.1 function due to aberrant N-glycosylation as a potential novel pathomechanism of SLE and ataxia in PMM2-CDG by using whole-cell patch-clamp, N-glycosylation blockade and mutagenesis. Nine SLE were identified. Neuroimages showed no signs of stroke. Comparison of characteristics between SLE positive versus negative patients' group showed no differences. Acute and chronic phenotypes of patients with PMM2-CDG or CACNA1A channelopathies show similarities. Hypoglycosylation of both CaV2.1 subunits (α1A and α2α) induced gain-of-function effects on channel gating that mirrored those reported for pathogenic CACNA1A mutations linked to FHM and ataxia. Unoccupied N-glycosylation site N283 at α1A contributes to a gain-of-function by lessening CaV2.1 inactivation. Hypoglycosylation of the α₂δ subunit also participates in the gain-of-function effect by promoting voltage-dependent opening of the CaV2.1 channel. CaV2.1 hypoglycosylation may cause ataxia and SLEs in PMM2-CDG patients. Aberrant CaV2.1 N-glycosylation as a novel pathomechanism in PMM2-CDG opens new therapeutic possibilities.
Keywords: CaV2.1 voltage-gated calcium channel; ataxia; cerebellum; congenital disorders of glycosylation; magentic resonance Imaging (MRI); stroke-like.
Conflict of interest statement
The authors report no conflicts of interest. We did not have any sponsor in any of the phases of the study. None of us, the authors, has received any payment to produce the manuscript.
Figures
References
-
- Feraco P., Mirabelli-Badenier M., Severino M., Alpigiani M.G., Di Rocco M., Biancheri R., Rossi A. The shrunken, bright cerebellum: A characteristic MRI finding in congenital disorders of glycosylation type 1a. AJNR Am. J. Neuroradiol. 2012;33:2062–2067. doi: 10.3174/ajnr.A3151. - DOI - PMC - PubMed
-
- Pérez-Dueñas B., García-Cazorla A., Pineda M., Poo P., Campistol J., Cusí V., Schollen E., Matthijs G., Grunewald S., Briones P., et al. Long-term evolution of eight Spanish patients with CDG type Ia: Typical and atypical manifestations. Eur. J. Paediatr. Neurol. 2009;13:444–451. doi: 10.1016/j.ejpn.2008.09.002. - DOI - PubMed
-
- Barone R., Carrozzi M., Parini R., Battini R., Martinelli D., Elia M., Spada M., Lilliu F., Ciana G., Burlina A., et al. A nationwide survey of PMM2-CDG in Italy: High frequency of a mild neurological variant associated with the L32R mutation. J. Neurol. 2015;262:154–164. doi: 10.1007/s00415-014-7549-7. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
