

Cholesterol-enriched membrane microdomains are needed for insulin signaling and proliferation in hepatic cells
- PMID: 29471671
- PMCID: PMC6109708
- DOI: 10.1152/ajpgi.00008.2018
Cholesterol-enriched membrane microdomains are needed for insulin signaling and proliferation in hepatic cells
Abstract
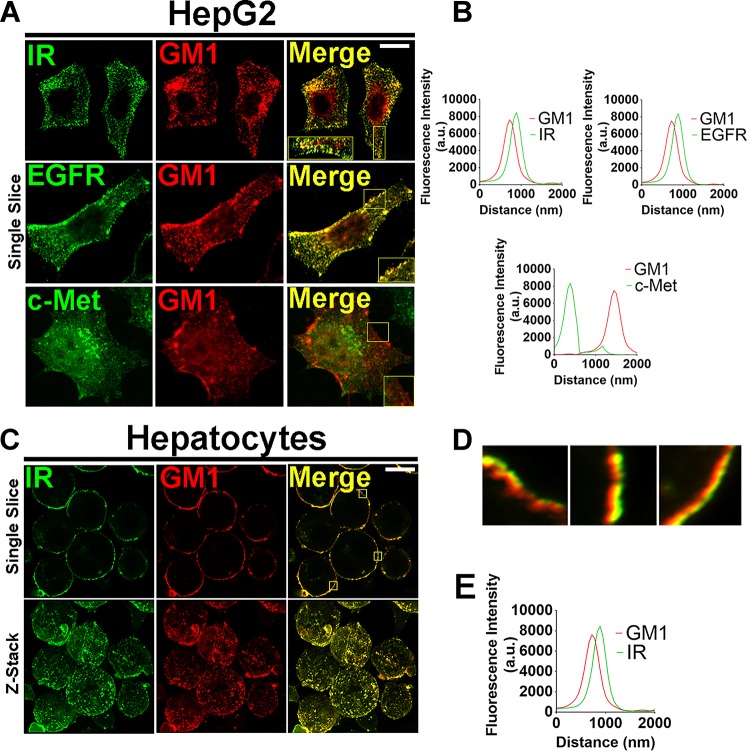
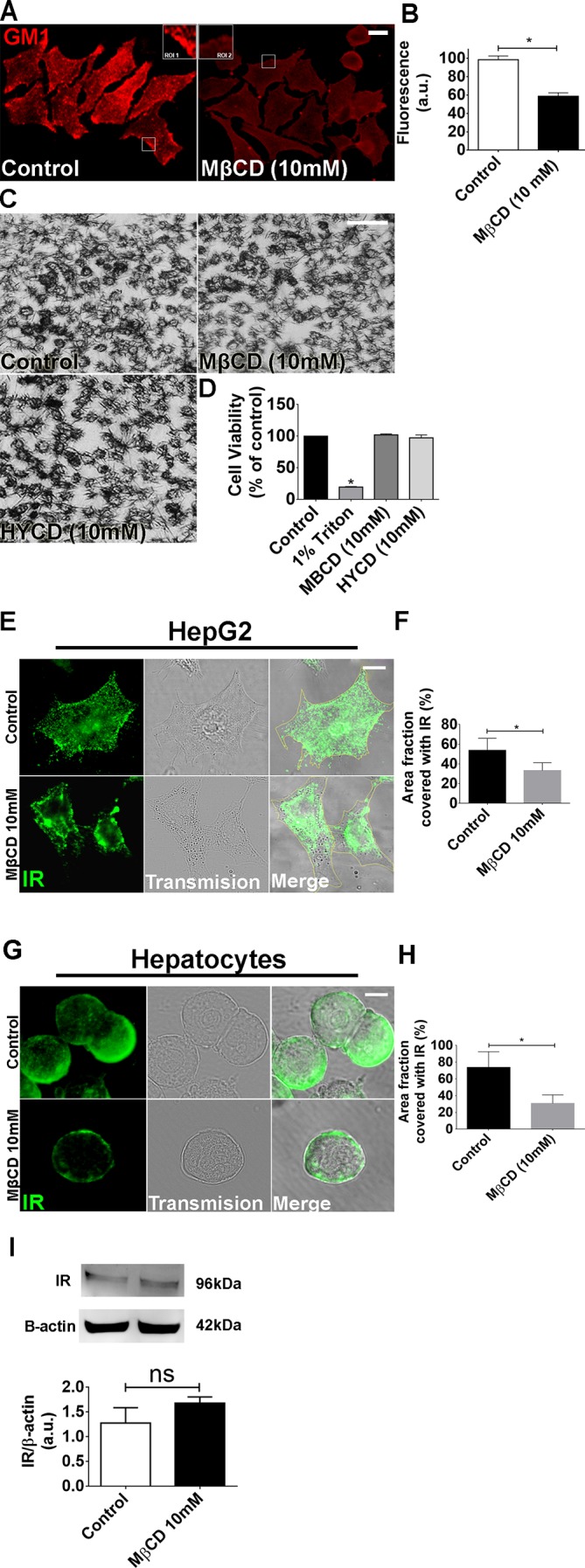
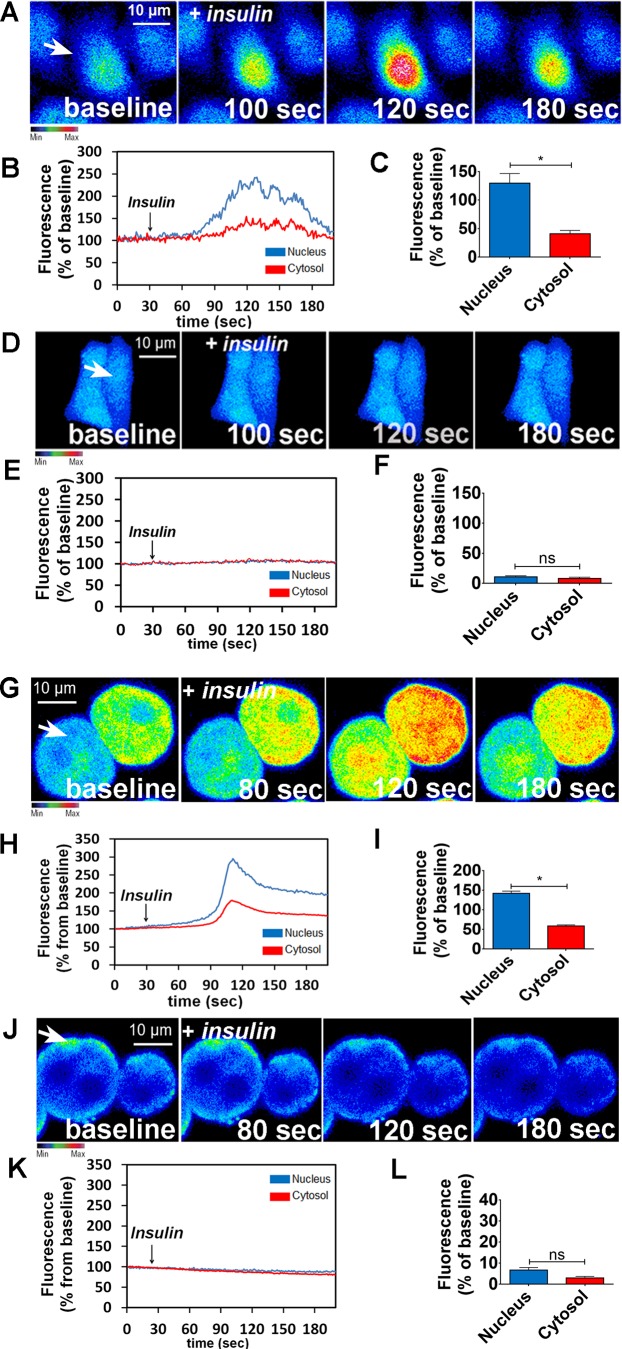
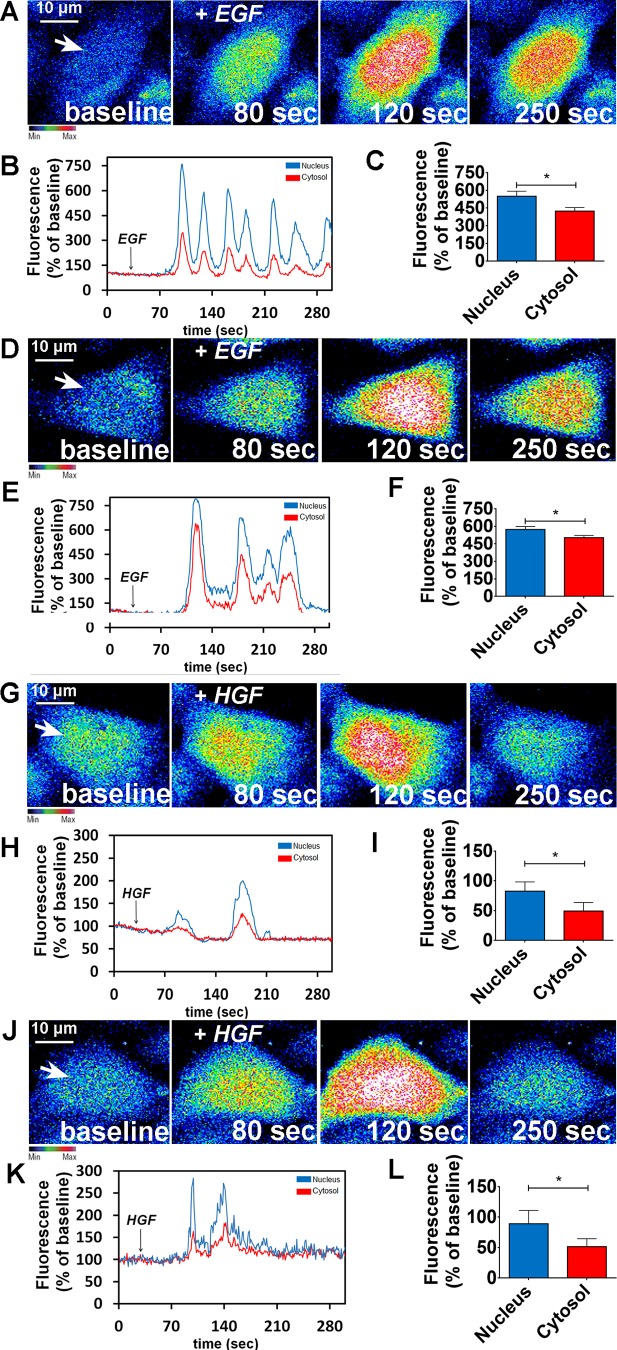
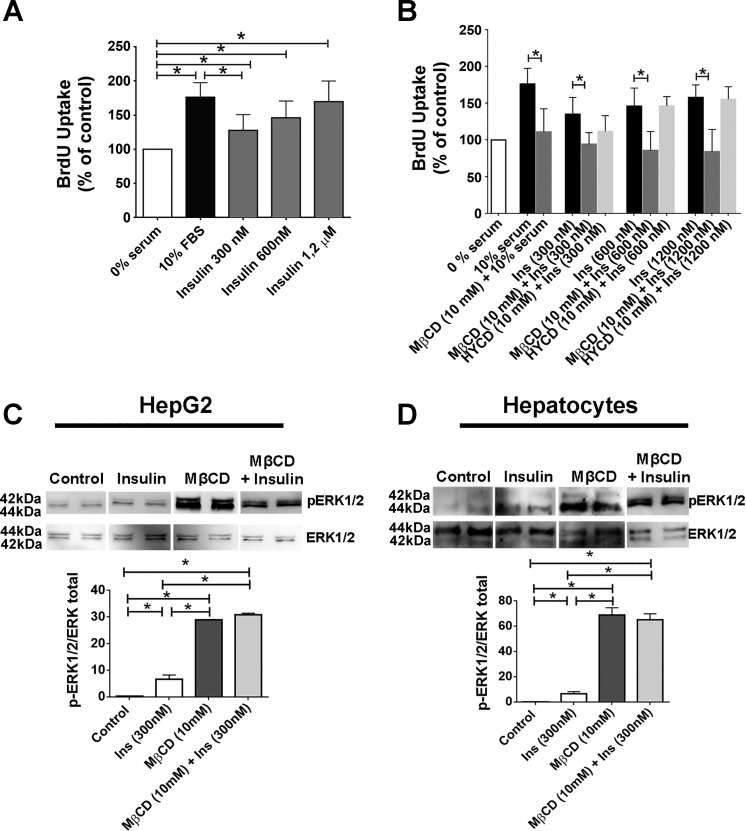
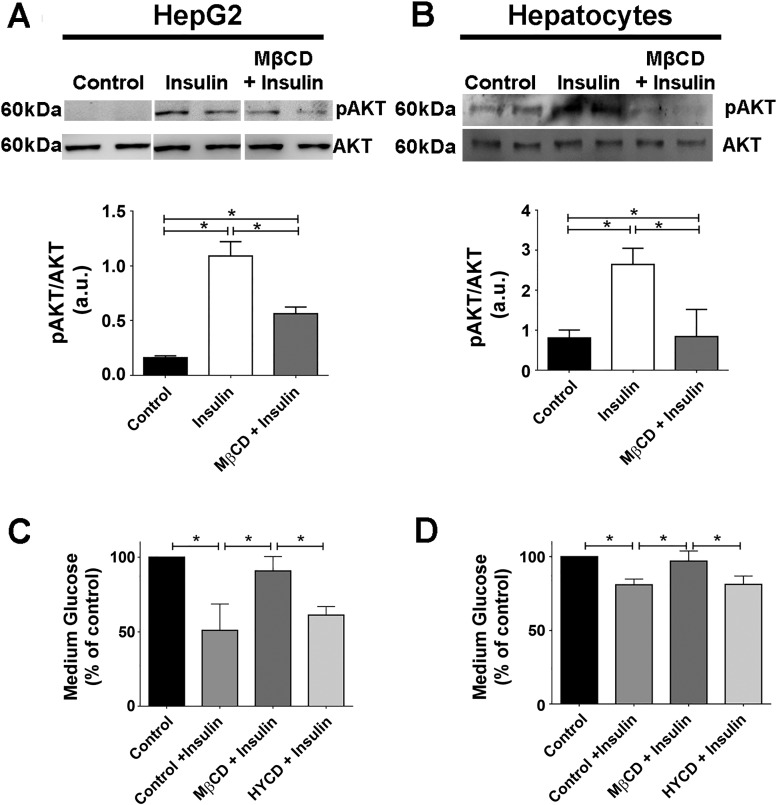
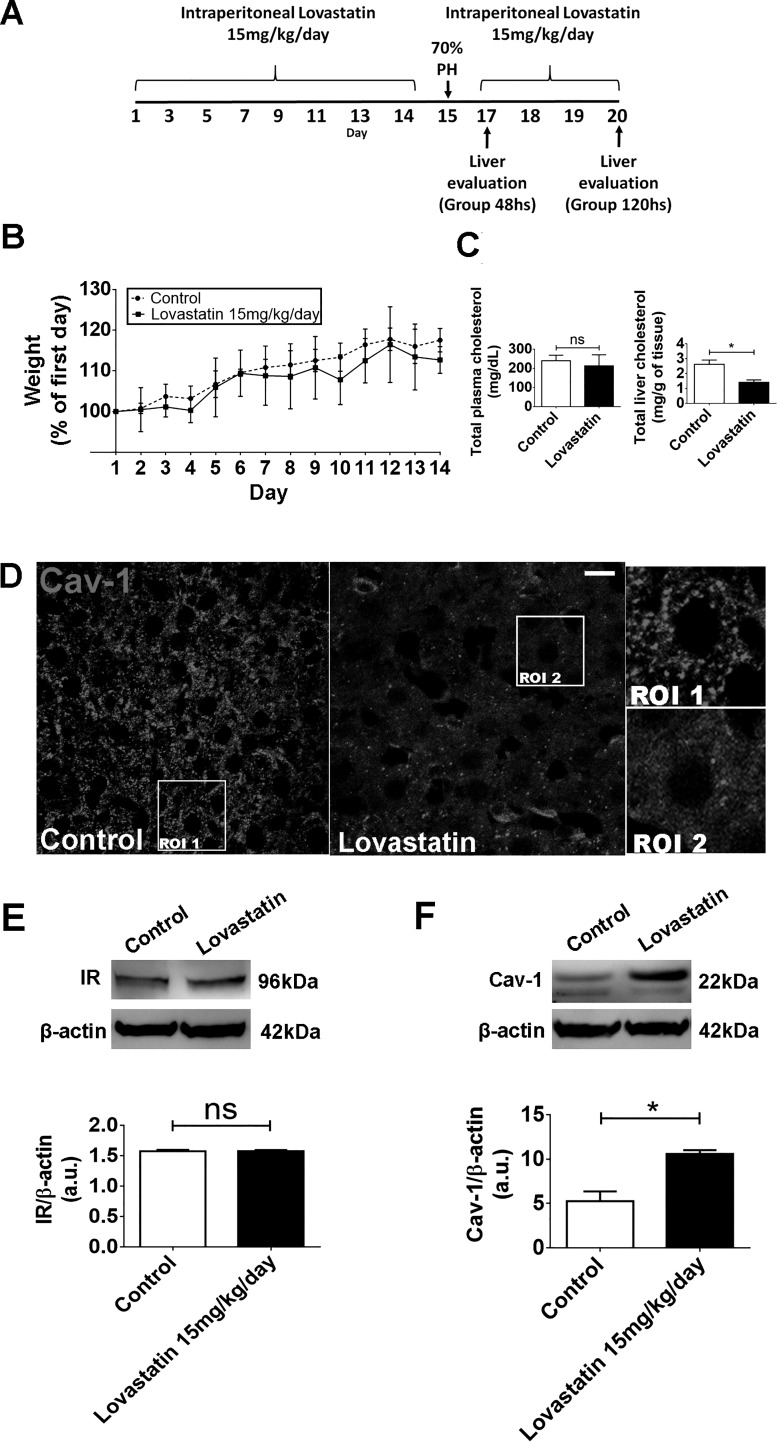
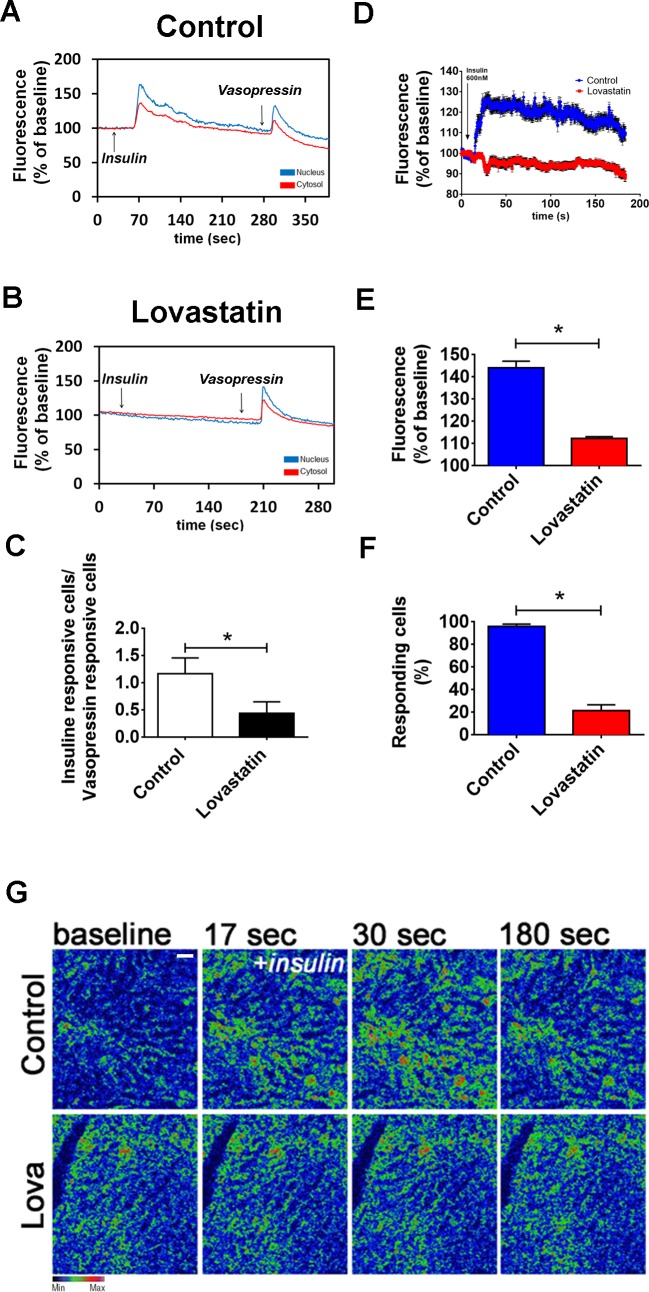
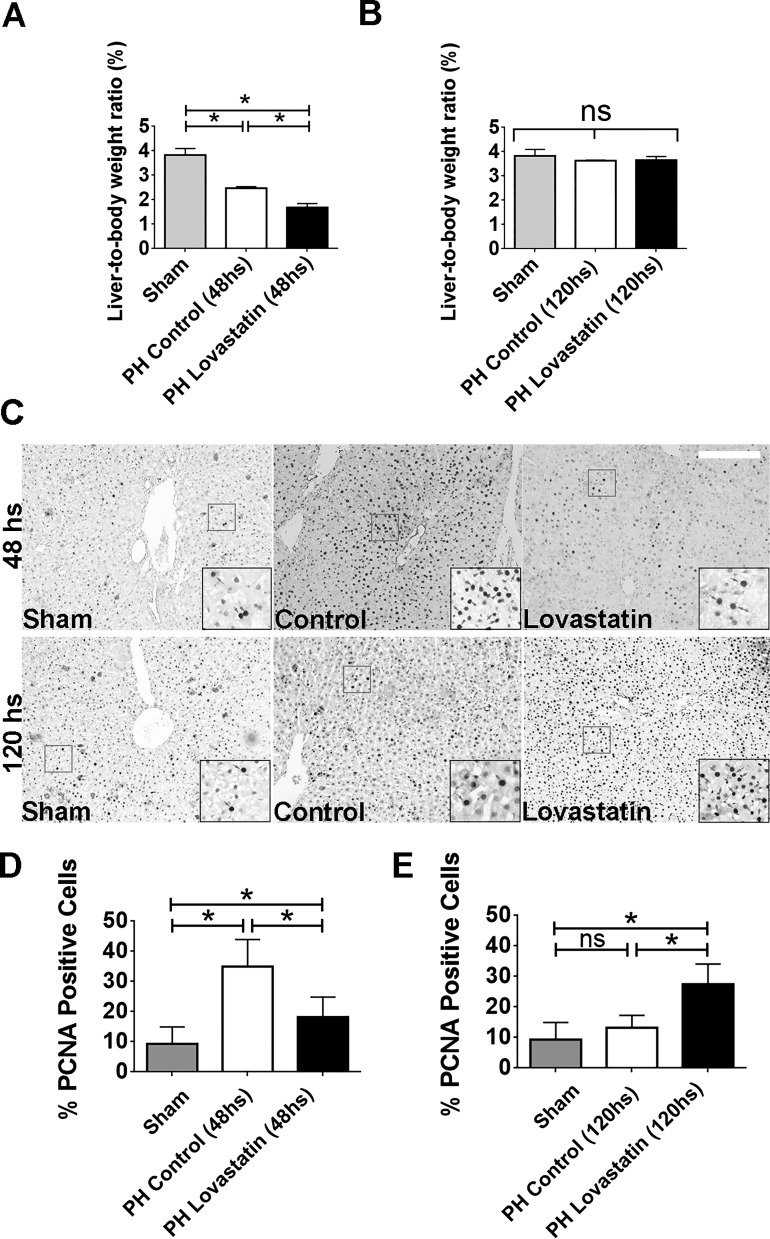
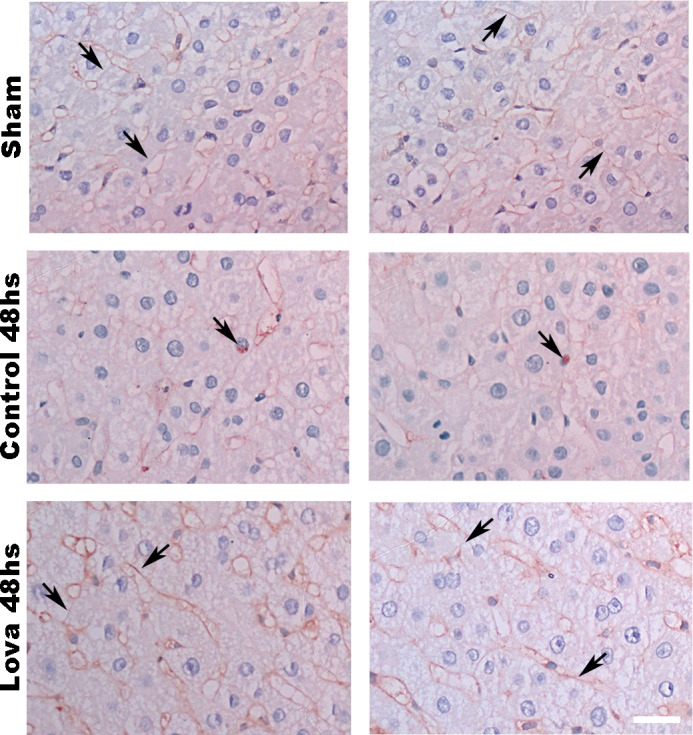
Hepatocyte proliferation during liver regeneration is a well-coordinated process regulated by the activation of several growth factor receptors, including the insulin receptor (IR). The IR can be localized in part to cholesterol-enriched membrane microdomains, but the role of such domains in insulin-mediated events in hepatocytes is not known. We investigated whether partitioning of IRs into cholesterol-enriched membrane rafts is important for the mitogenic effects of insulin in the hepatic cells. IR and lipid rafts were labeled in HepG2 cells and primary rat hepatocytes. Membrane cholesterol was depleted in vitro with metyl-β-cyclodextrin (MβCD) and in vivo with lovastatin. Insulin-induced calcium (Ca2+) signals studies were examined in HepG2 cells and in freshly isolated rat hepatocytes as well as in whole liver in vivo by intravital confocal imaging. Liver regeneration was studied by 70% partial hepatectomy (PH), and hepatocyte proliferation was assessed by PCNA staining. A subpopulation of IR was found in membrane microdomains enriched in cholesterol. Depletion of cholesterol from plasma membrane resulted in redistribution of the IR along the cells, which was associated with impaired insulin-induced nuclear Ca2+ signals, a signaling event that regulates hepatocyte proliferation. Cholesterol depletion also led to ERK1/2 hyper-phosphorylation. Lovastatin administration to rats decreased hepatic cholesterol content, disrupted lipid rafts and decreased insulin-induced Ca2+ signaling in hepatocytes, and delayed liver regeneration after PH. Therefore, membrane cholesterol content and lipid rafts integrity showed to be important for the proliferative effects of insulin in hepatic cells. NEW & NOTEWORTHY One of insulin's actions is to stimulate liver regeneration. Here we show that a subpopulation of insulin receptors is in a specialized cholesterol-enriched region of the cell membrane and this subfraction is important for insulin's proliferative effects.
Keywords: calcium signaling; hepatocytes; insulin signaling; lipid rafts; liver regeneration.
Figures
References
-
- Amaya MJ, Oliveira AG, Guimarães ES, Casteluber MC, Carvalho SM, Andrade LM, Pinto MC, Mennone A, Oliveira CA, Resende RR, Menezes GB, Nathanson MH, Leite MF. The insulin receptor translocates to the nucleus to regulate cell proliferation in liver. Hepatology 59: 274–283, 2014. doi:10.1002/hep.26609. - DOI - PMC - PubMed
-
- Berridge MV, Tan AS. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch Biochem Biophys 303: 474–482, 1993. doi:10.1006/abbi.1993.1311. - DOI - PubMed
-
- Björkhem-Bergman L, Acimovic J, Torndal UB, Parini P, Eriksson LC. Lovastatin prevents carcinogenesis in a rat model for liver cancer. Effects of ubiquinone supplementation. Anticancer Res 30: 1105–1112, 2010. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous