

A maximum likelihood algorithm for reconstructing 3D structures of human chromosomes from chromosomal contact data
- PMID: 29471801
- PMCID: PMC5824572
- DOI: 10.1186/s12864-018-4546-8
A maximum likelihood algorithm for reconstructing 3D structures of human chromosomes from chromosomal contact data
Abstract
Background: The development of chromosomal conformation capture techniques, particularly, the Hi-C technique, has made the analysis and study of the spatial conformation of a genome an important topic in bioinformatics and computational biology. Aided by high-throughput next generation sequencing techniques, the Hi-C technique can generate genome-wide, large-scale intra- and inter-chromosomal interaction data capable of describing in details the spatial interactions within a genome. These data can be used to reconstruct 3D structures of chromosomes that can be used to study DNA replication, gene regulation, genome interaction, genome folding, and genome function.
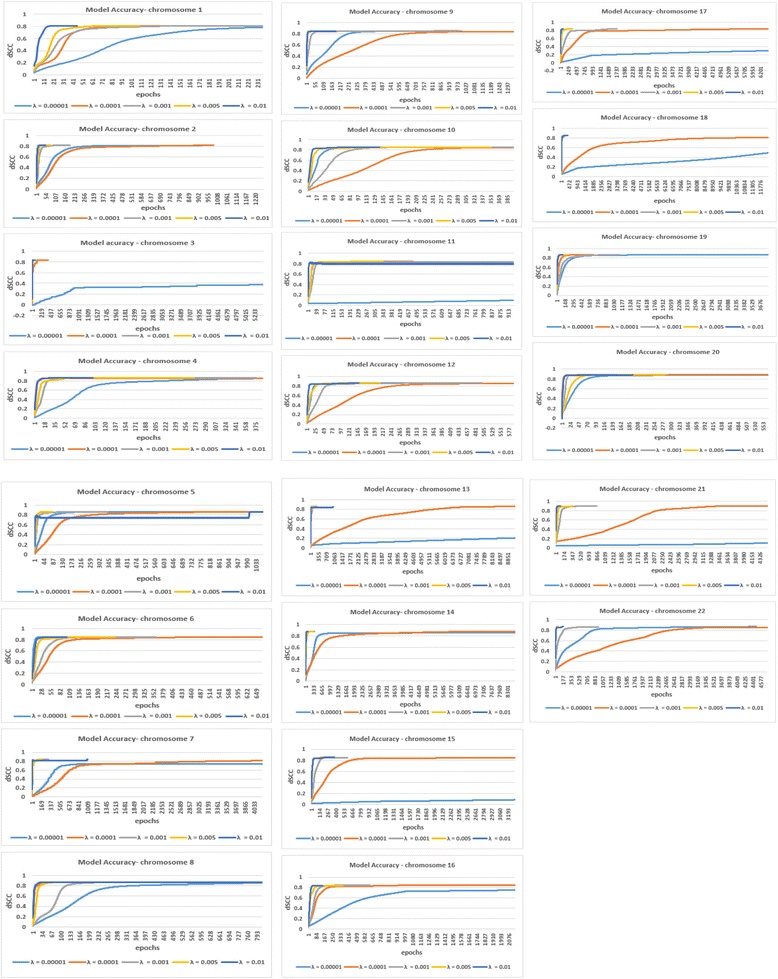
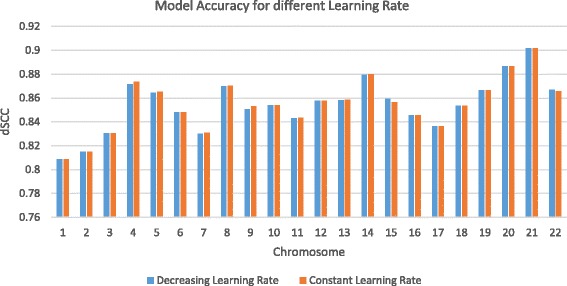
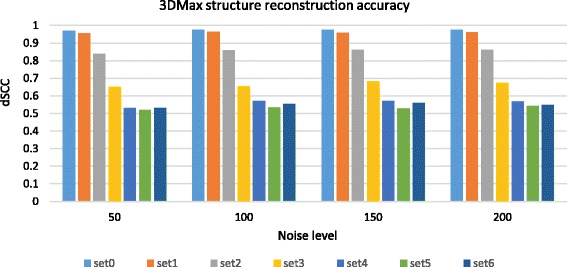
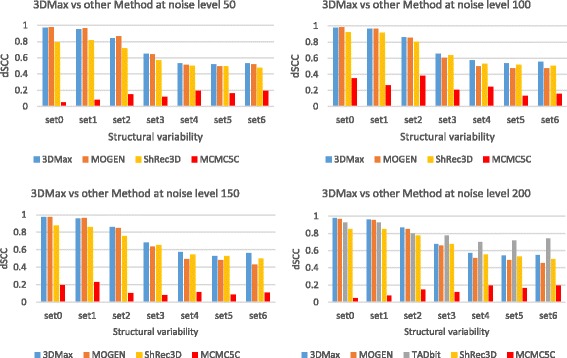
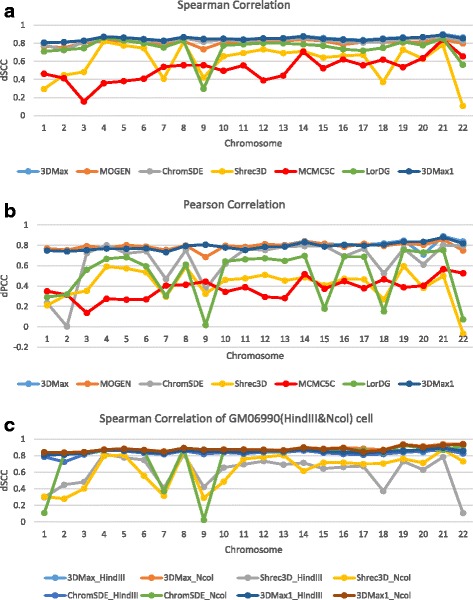
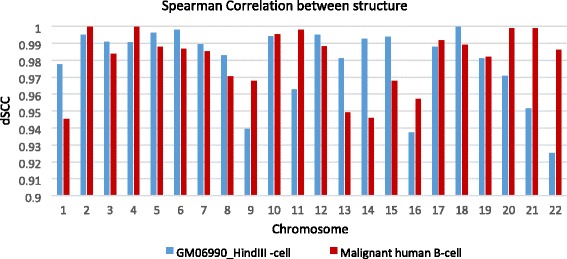
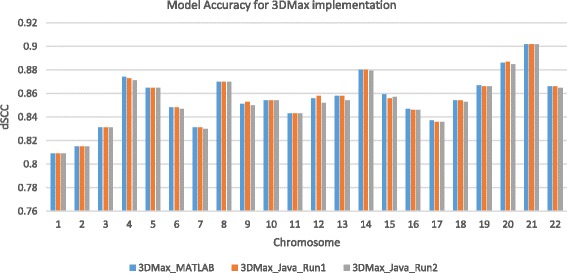
Results: Here, we introduce a maximum likelihood algorithm called 3DMax to construct the 3D structure of a chromosome from Hi-C data. 3DMax employs a maximum likelihood approach to infer the 3D structures of a chromosome, while automatically re-estimating the conversion factor (α) for converting Interaction Frequency (IF) to distance. Our results show that the models generated by 3DMax from a simulated Hi-C dataset match the true models better than most of the existing methods. 3DMax is more robust to structural variability and noise. Compared on a real Hi-C dataset, 3DMax constructs chromosomal models that fit the data better than most methods, and it is faster than all other methods. The models reconstructed by 3DMax were consistent with fluorescent in situ hybridization (FISH) experiments and existing knowledge about the organization of human chromosomes, such as chromosome compartmentalization.
Conclusions: 3DMax is an effective approach to reconstructing 3D chromosomal models. The results, and the models generated for the simulated and real Hi-C datasets are available here: http://sysbio.rnet.missouri.edu/bdm_download/3DMax/ . The source code is available here: https://github.com/BDM-Lab/3DMax . A short video demonstrating how to use 3DMax can be found here: https://youtu.be/ehQUFWoHwfo .
Keywords: 3D chromosome structure; 3D genome; Chromosome conformation capture; Gradient ascent; Hi-C.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
