

Idiopathic pulmonary fibrosis: pathogenesis and management
- PMID: 29471816
- PMCID: PMC5824456
- DOI: 10.1186/s12931-018-0730-2
Idiopathic pulmonary fibrosis: pathogenesis and management
Abstract
Background: Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive disease characterized by the aberrant accumulation of fibrotic tissue in the lungs parenchyma, associated with significant morbidity and poor prognosis. This review will present the substantial advances achieved in the understanding of IPF pathogenesis and in the therapeutic options that can be offered to patients, and will address the issues regarding diagnosis and management that are still open.
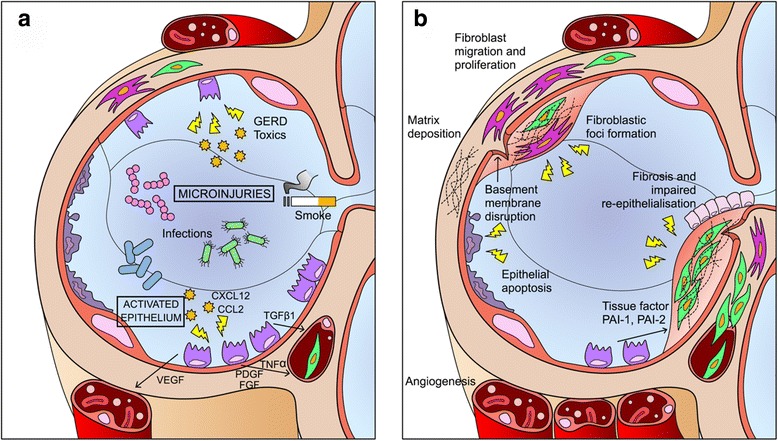
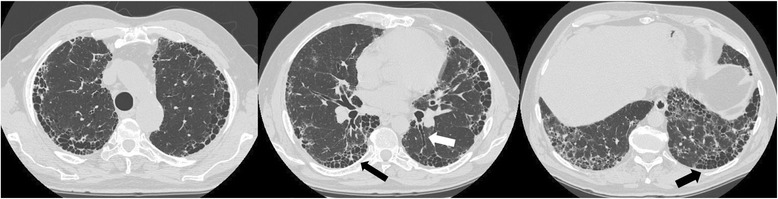
Main body: Over the last two decades much has been clarified about the pathogenic pathways underlying the development and progression of the lung scarring in IPF. Sustained alveolar epithelial micro-injury and activation has been recognised as the trigger of several biological events of disordered repair occurring in genetically susceptible ageing individuals. Despite multidisciplinary team discussion has demonstrated to increase diagnostic accuracy, patients can still remain unclassified when the current diagnostic criteria are strictly applied, requiring the identification of a Usual Interstitial Pattern either on high-resolution computed tomography scan or lung biopsy. Outstanding achievements have been made in the management of these patients, as nintedanib and pirfenidone consistently proved to reduce the rate of progression of the fibrotic process. However, many uncertainties still lie in the correct use of these drugs, ranging from the initial choice of the drug, the appropriate timing for treatment and the benefit-risk ratio of a combined treatment regimen. Several novel compounds are being developed in the perspective of a more targeted therapeutic approach; in the meantime, the supportive care of these patients and their carers should be appropriately prioritized, and greater efforts should be made toward the prompt identification and management of relevant comorbidities.
Conclusions: Building on the advances in the understanding of IPF pathobiology, the further investigation of the role of gene variants, epigenetic alterations and other molecular biomarkers reflecting disease activity and behaviour will hopefully enable earlier and more confident diagnosis, improve disease phenotyping and support the development of novel agents for personalized treatment of IPF.
Keywords: Diagnosis; Idiopathic pulmonary fibrosis; Interstitial lung disease; Management; Nintedanib; Pathogenesis; Pirfenidone; Treatment.
Conflict of interest statement
Authors’ information
Optional.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
GS and FV report personal fees from Boehringer Ingelheim outside the submitted work. BI, MC and MO declare that they have no competing interests.LR reports personal fees from Medimmune, personal fees from Biogen, personal fees from Sanofi-Aventis, personal fees from Takeda, personal fees from ImmuneWorks, personal fees from Shionogi, personal fees from Cipla, personal fees from Pliants Therapeutics, personal fees from Boehringer Ingelheim, personal fees from Roche.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Taskar VS. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc. 2006;3:293–8. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
