

Patient-derived organoids model treatment response of metastatic gastrointestinal cancers
- PMID: 29472484
- PMCID: PMC6112415
- DOI: 10.1126/science.aao2774
Patient-derived organoids model treatment response of metastatic gastrointestinal cancers
Abstract
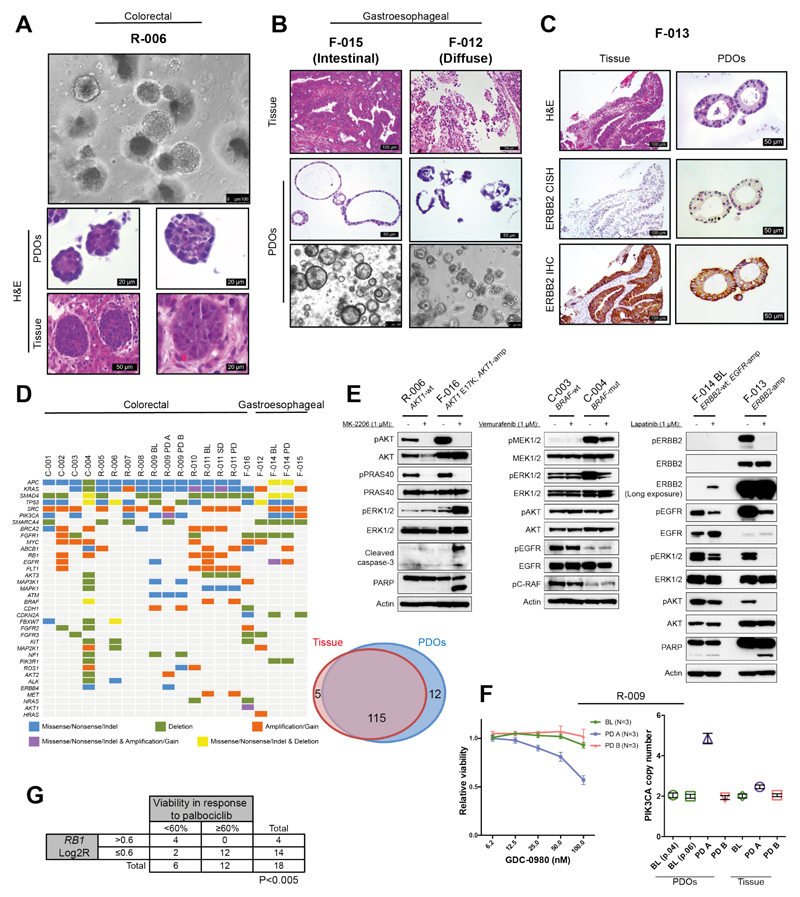
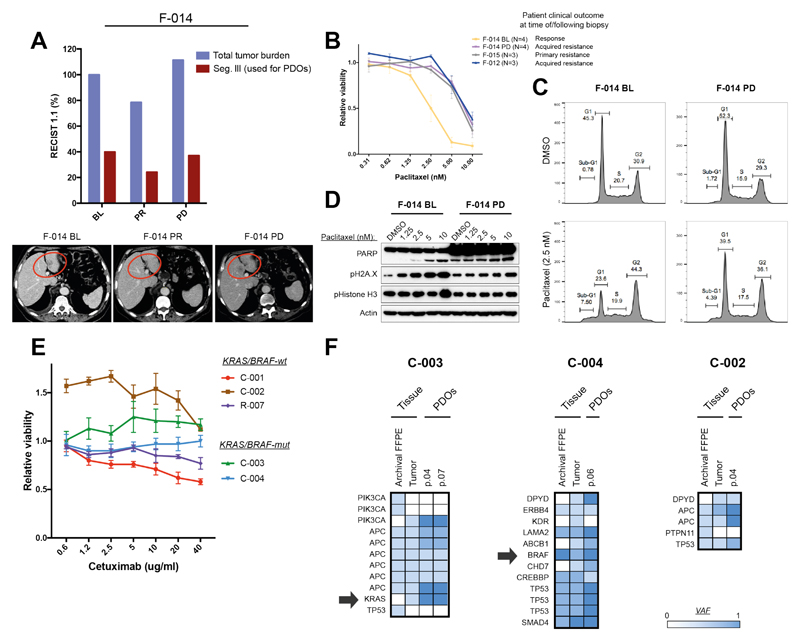
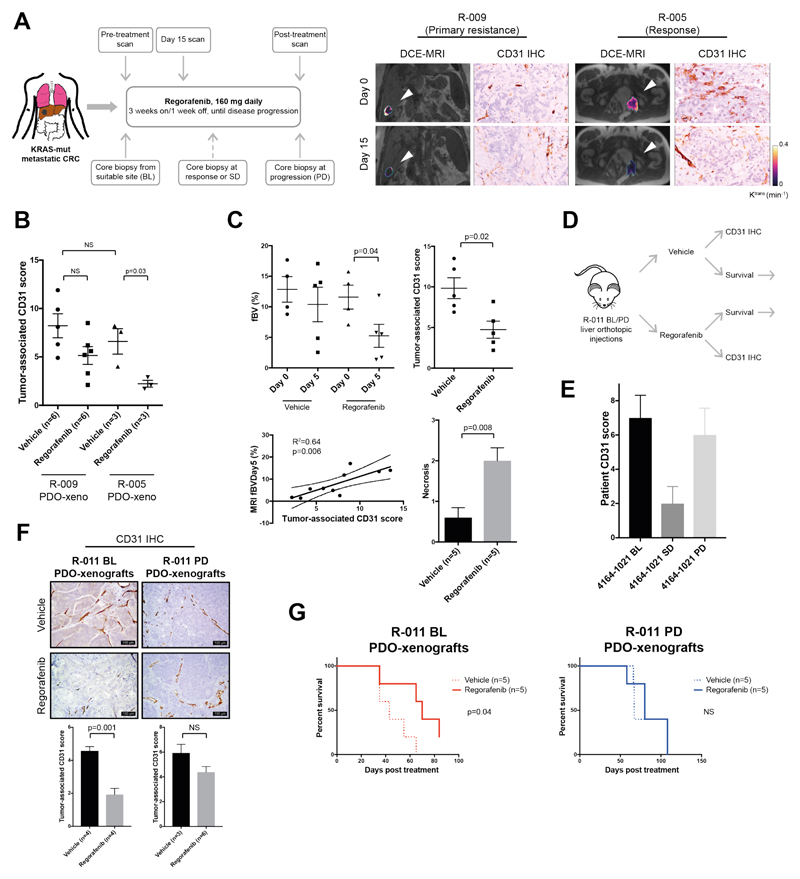
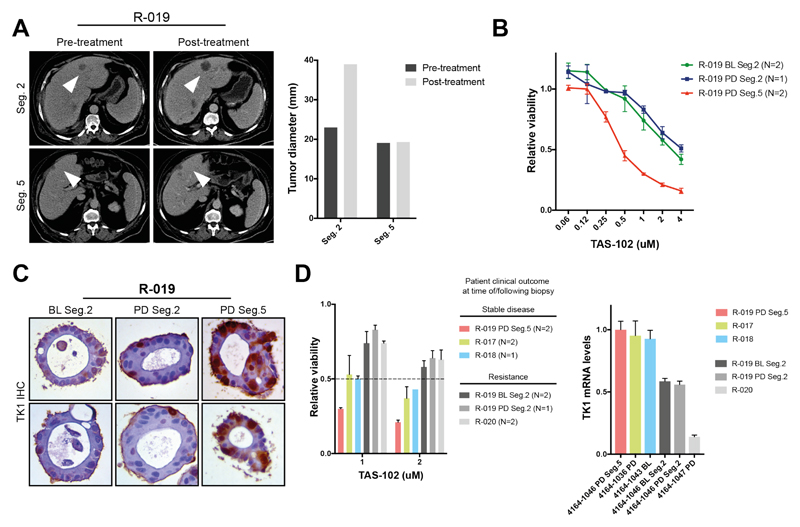
Patient-derived organoids (PDOs) have recently emerged as robust preclinical models; however, their potential to predict clinical outcomes in patients has remained unclear. We report on a living biobank of PDOs from metastatic, heavily pretreated colorectal and gastroesophageal cancer patients recruited in phase 1/2 clinical trials. Phenotypic and genotypic profiling of PDOs showed a high degree of similarity to the original patient tumors. Molecular profiling of tumor organoids was matched to drug-screening results, suggesting that PDOs could complement existing approaches in defining cancer vulnerabilities and improving treatment responses. We compared responses to anticancer agents ex vivo in organoids and PDO-based orthotopic mouse tumor xenograft models with the responses of the patients in clinical trials. Our data suggest that PDOs can recapitulate patient responses in the clinic and could be implemented in personalized medicine programs.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
All other authors declare no conflict of interest.
Figures
Comment in
-
Organoids May Point to Best Therapy.Cancer Discov. 2018 May;8(5):524. doi: 10.1158/2159-8290.CD-NB2018-029. Epub 2018 Mar 13. Cancer Discov. 2018. PMID: 29535134
-
Gastrointestinal cancer: Organoids predict clinical responses.Nat Rev Gastroenterol Hepatol. 2018 Apr;15(4):189. doi: 10.1038/nrgastro.2018.25. Epub 2018 Mar 14. Nat Rev Gastroenterol Hepatol. 2018. PMID: 29535457 No abstract available.
-
Organoids test drug response.Nat Cell Biol. 2018 Jun;20(6):634. doi: 10.1038/s41556-018-0116-1. Nat Cell Biol. 2018. PMID: 29784911 No abstract available.
References
-
- Voest EE, Bernards R. DNA-guided precision medicine for cancer: a case of irrational exuberance? Cancer Discovery. 2016;6(2):130–132. - PubMed
-
- Byrne AT, et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat Rev Cancer. 2017;17(4):254–268. - PubMed
-
- Clohessy JG, Pandolfi PP. Mouse hospital and co-clinical trial project--from bench to bedside. Nat Rev Clin Oncol. 2015;12(8):491–498. - PubMed
-
- Bredenoord AL, Clevers H, Knoblich JA. Human tissues in a dish: The research and ethical implications of organoid technology. Science. 2017;355(6322):eaaf9414. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
