

Exploiting genetic variation to uncover rules of transcription factor binding and chromatin accessibility
- PMID: 29472540
- PMCID: PMC5823854
- DOI: 10.1038/s41467-018-03082-6
Exploiting genetic variation to uncover rules of transcription factor binding and chromatin accessibility
Abstract
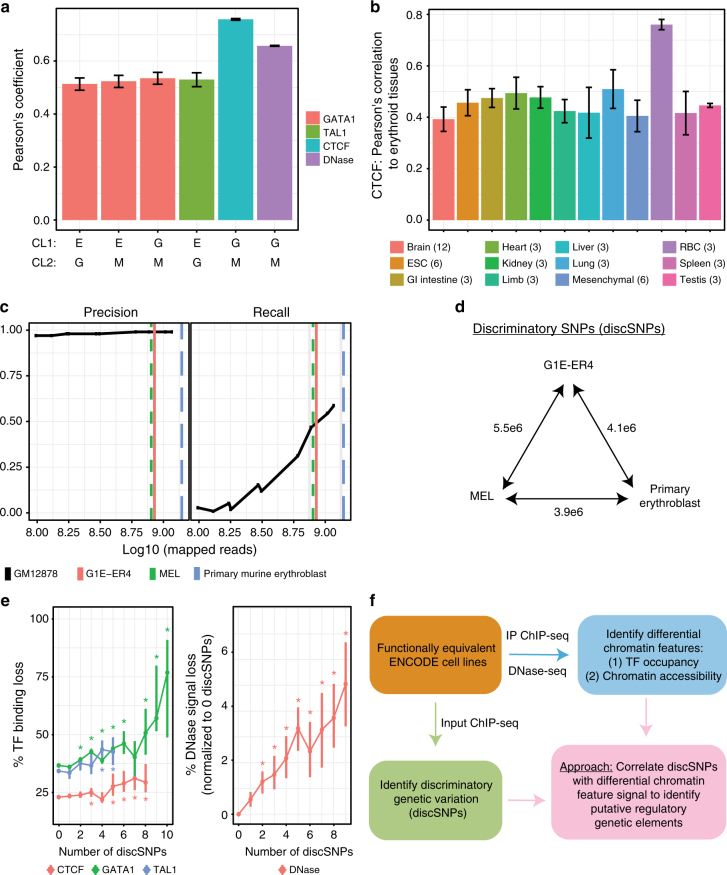
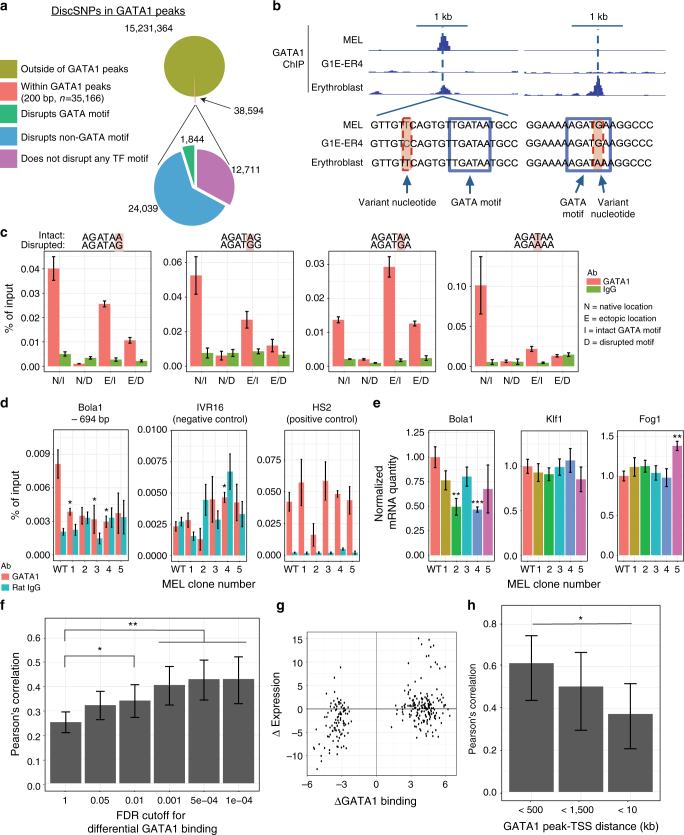
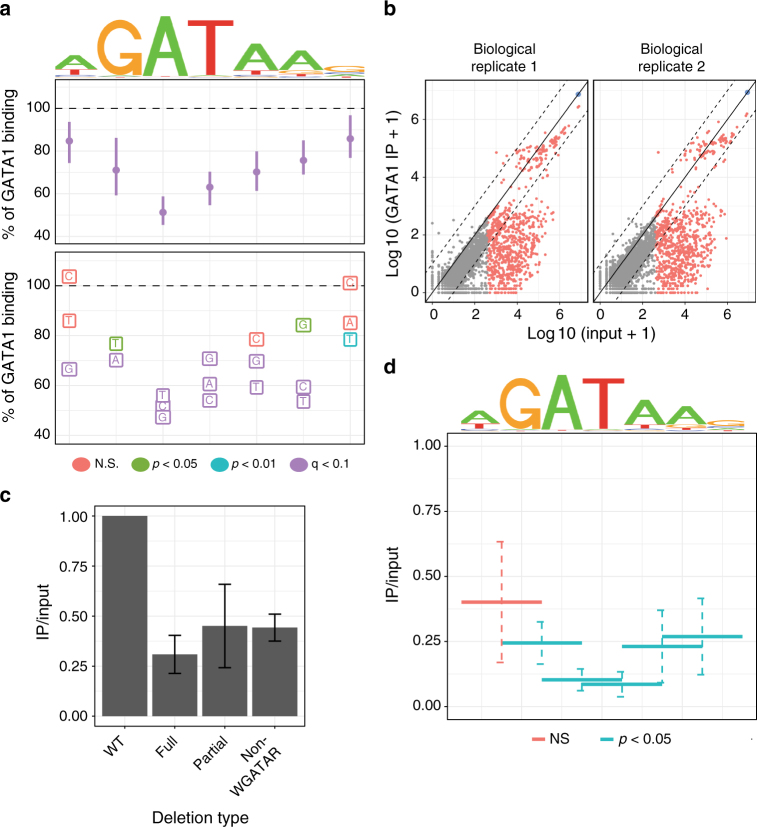
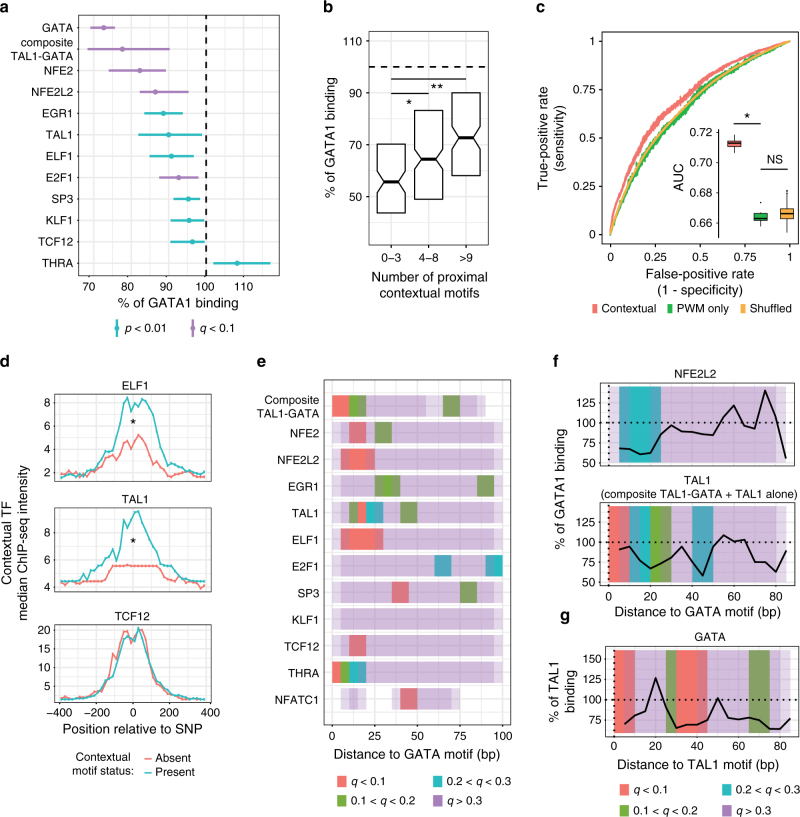
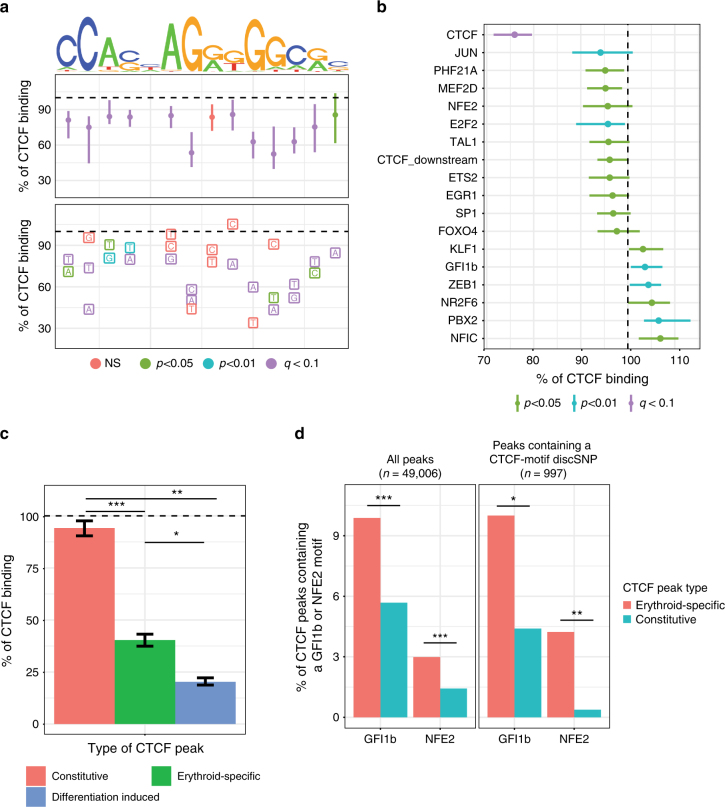
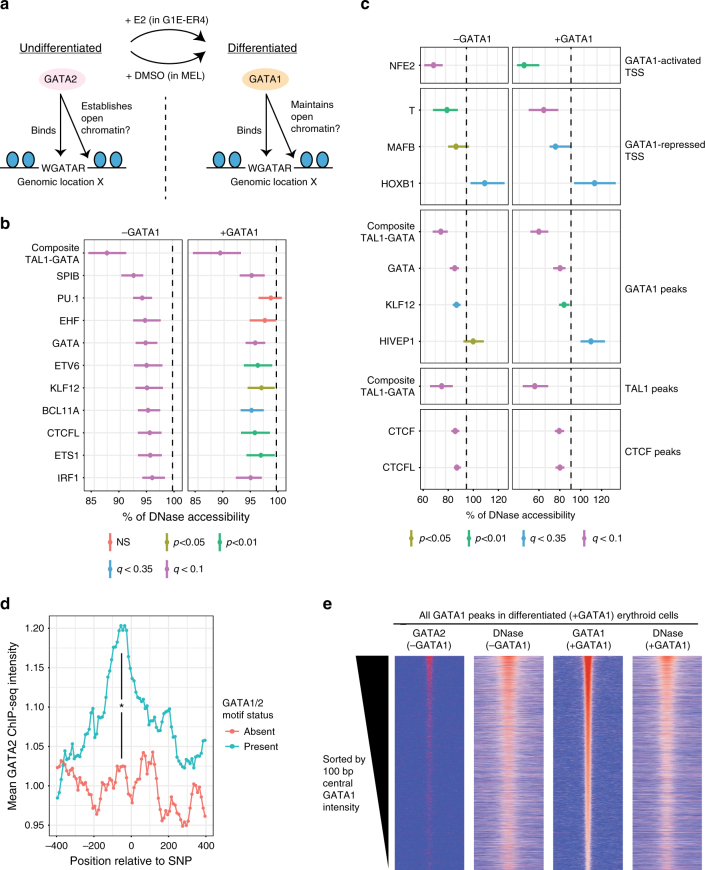
Single-nucleotide variants that underlie phenotypic variation can affect chromatin occupancy of transcription factors (TFs). To delineate determinants of in vivo TF binding and chromatin accessibility, we introduce an approach that compares ChIP-seq and DNase-seq data sets from genetically divergent murine erythroid cell lines. The impact of discriminatory single-nucleotide variants on TF ChIP signal enables definition at single base resolution of in vivo binding characteristics of nuclear factors GATA1, TAL1, and CTCF. We further develop a facile complementary approach to more deeply test the requirements of critical nucleotide positions for TF binding by combining CRISPR-Cas9-mediated mutagenesis with ChIP and targeted deep sequencing. Finally, we extend our analytical pipeline to identify nearby contextual DNA elements that modulate chromatin binding by these three TFs, and to define sequences that impact kb-scale chromatin accessibility. Combined, our approaches reveal insights into the genetic basis of TF occupancy and their interplay with chromatin features.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
