

A motif within the armadillo repeat of Parkinson's-linked LRRK2 interacts with FADD to hijack the extrinsic death pathway
- PMID: 29472595
- PMCID: PMC5823876
- DOI: 10.1038/s41598-018-21931-8
A motif within the armadillo repeat of Parkinson's-linked LRRK2 interacts with FADD to hijack the extrinsic death pathway
Abstract
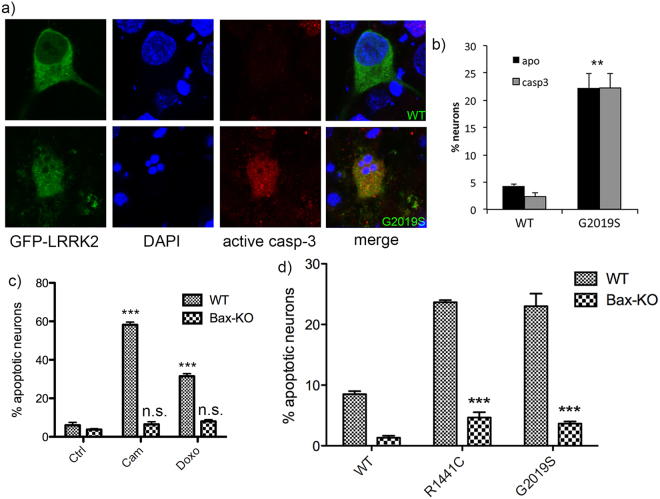
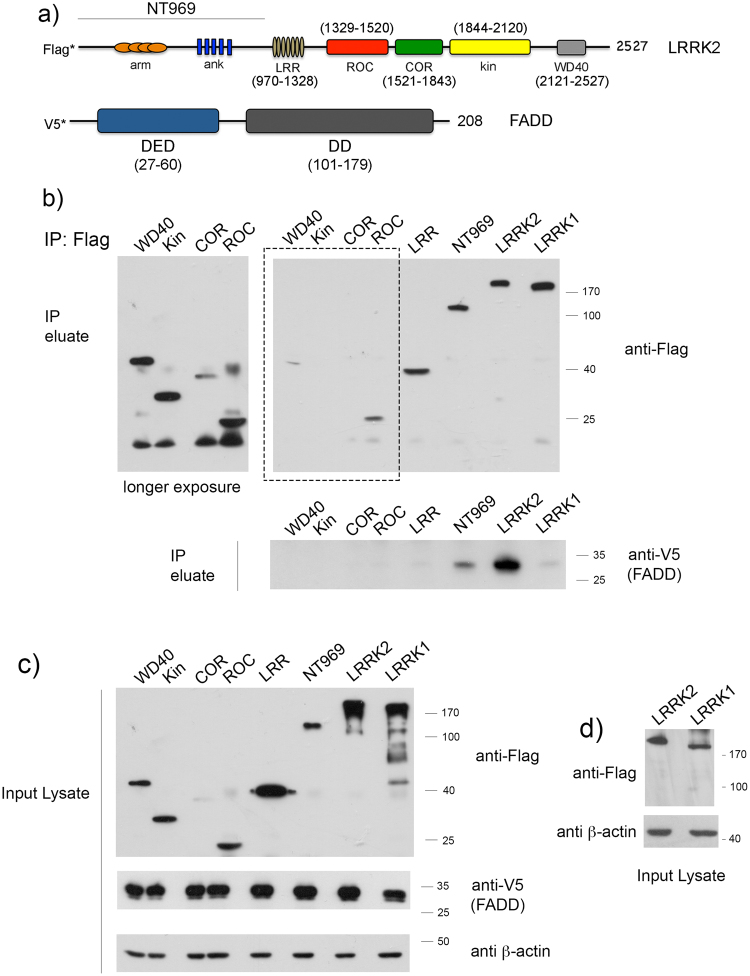
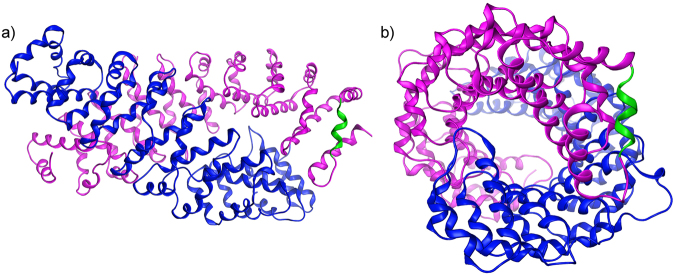
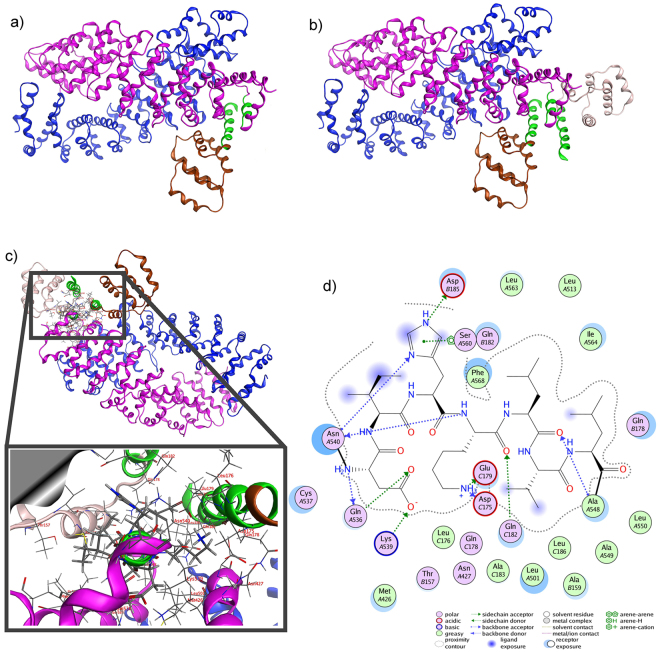
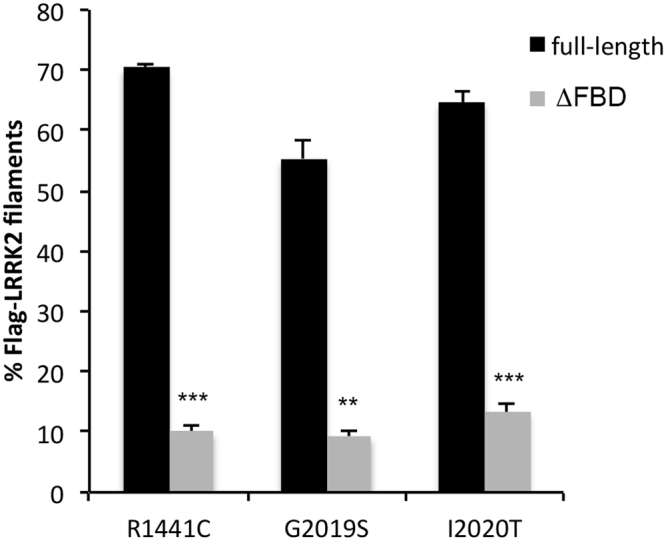
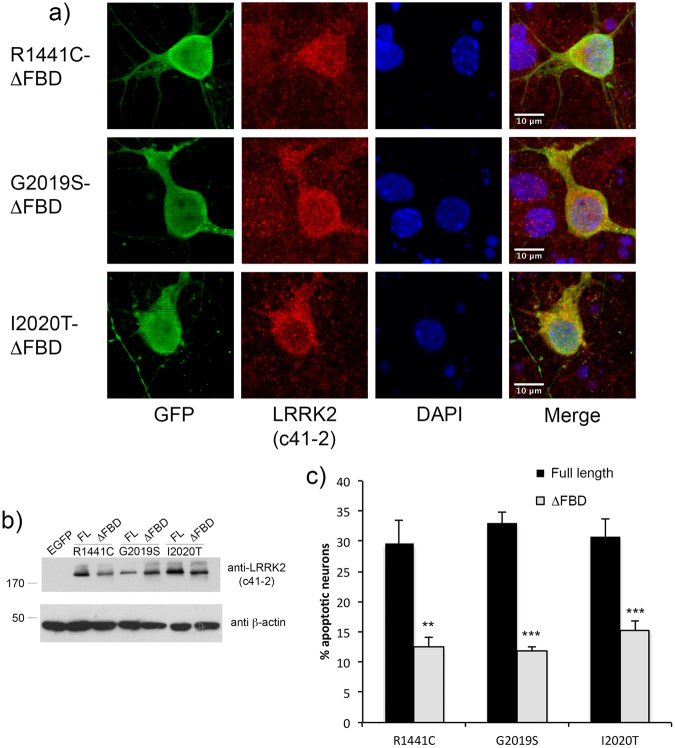
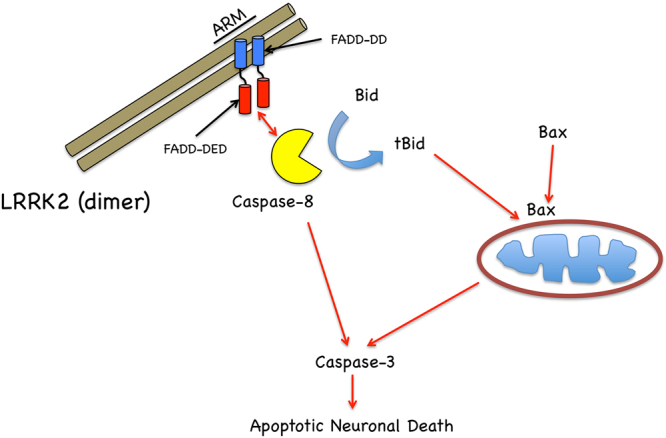
In experimental models, both in vivo and cellular, over-expression of Parkinson's linked mutant leucine-rich repeat kinase 2 (LRRK2) is sufficient to induce neuronal death. While several cell death associated proteins have been linked to LRRK2, either as protein interactors or as putative substrates, characterization of the neuronal death cascade remains elusive. In this study, we have mapped for the first time the domain within LRRK2 that mediates the interaction with FADD, thereby activating the molecular machinery of the extrinsic death pathway. Using homology modeling and molecular docking approaches, we have identified a critical motif within the N-terminal armadillo repeat region of LRRK2. Moreover, we show that co-expression of fragments of LRRK2 that contain the FADD binding motif, or deletion of this motif itself, blocks the interaction with FADD, and is neuroprotective. We further demonstrate that downstream of FADD, the mitochondrial proteins Bid and Bax are recruited to the death cascade and are necessary for neuronal death. Our work identifies multiple novel points within neuronal death signaling pathways that could potentially be targeted by candidate therapeutic strategies and highlight how the extrinsic pathway can be activated intracellularly in a pathogenic context.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Ho CC, Rideout HJ, Ribe E, Troy CM, Dauer WT. The Parkinson disease protein leucine-rich repeat kinase 2 transduces death signals via Fas-associated protein with death domain and caspase-8 in a cellular model of neurodegeneration. J Neurosci. 2009;29:1011–1016. doi: 10.1523/JNEUROSCI.5175-08.2009. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
