

scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells
- PMID: 29472610
- PMCID: PMC5823944
- DOI: 10.1038/s41467-018-03149-4
scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells
Abstract
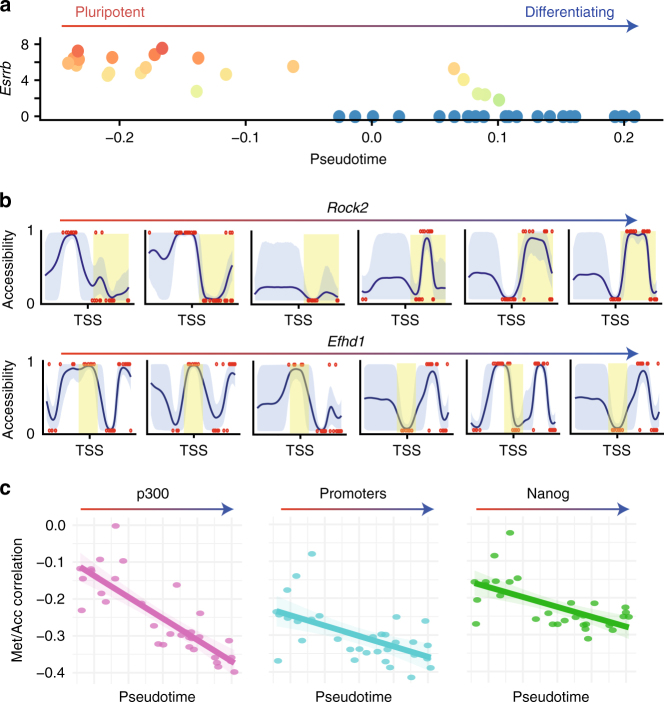
Parallel single-cell sequencing protocols represent powerful methods for investigating regulatory relationships, including epigenome-transcriptome interactions. Here, we report a single-cell method for parallel chromatin accessibility, DNA methylation and transcriptome profiling. scNMT-seq (single-cell nucleosome, methylation and transcription sequencing) uses a GpC methyltransferase to label open chromatin followed by bisulfite and RNA sequencing. We validate scNMT-seq by applying it to differentiating mouse embryonic stem cells, finding links between all three molecular layers and revealing dynamic coupling between epigenomic layers during differentiation.
Conflict of interest statement
W.R. is a consultant and shareholder of Cambridge Epigenetix. The remaining authors declare no competing financial interests.
Figures
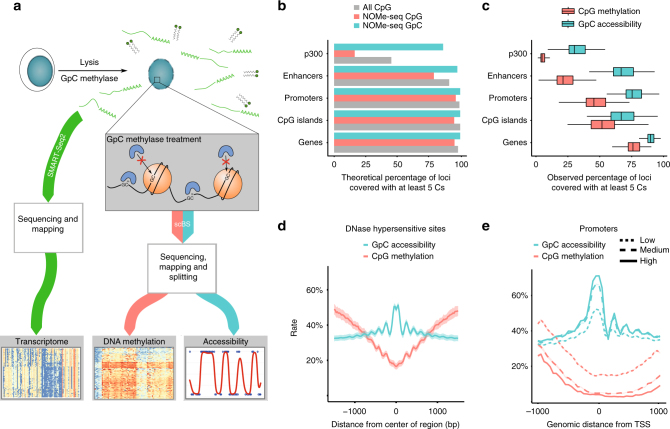
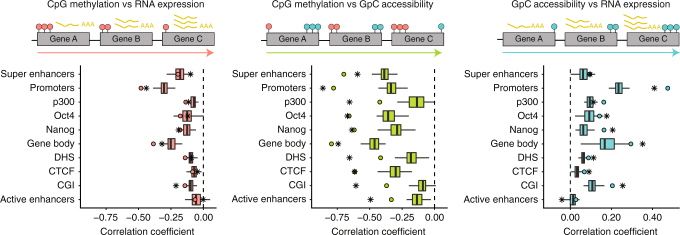
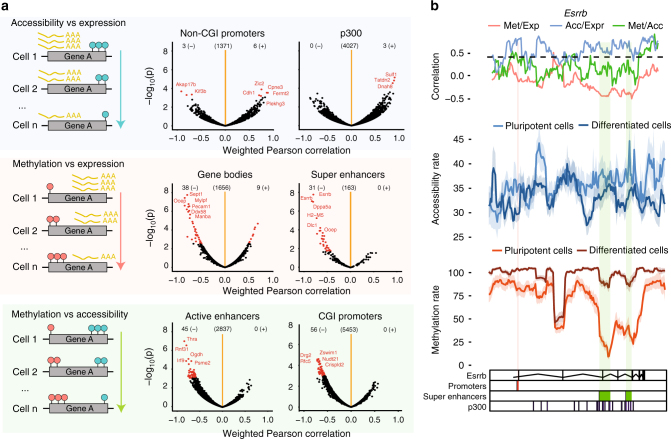
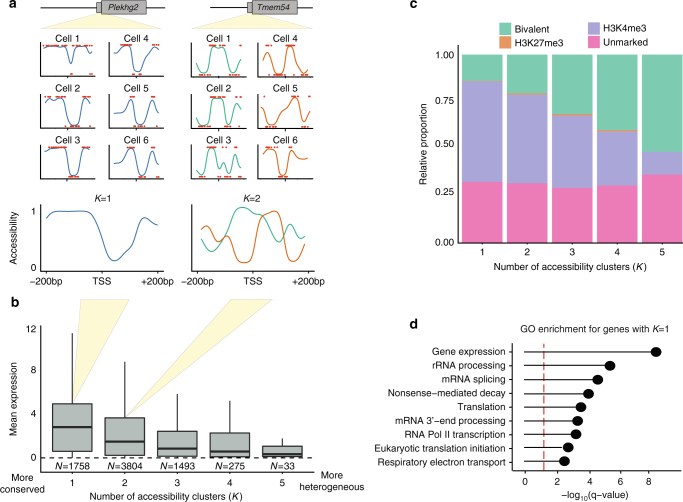
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
