

PennDiff: detecting differential alternative splicing and transcription by RNA sequencing
- PMID: 29474557
- PMCID: PMC6041879
- DOI: 10.1093/bioinformatics/bty097
PennDiff: detecting differential alternative splicing and transcription by RNA sequencing
Abstract
Motivation: Alternative splicing and alternative transcription are a major mechanism for generating transcriptome diversity. Differential alternative splicing and transcription (DAST), which describe different usage of transcript isoforms across different conditions, can complement differential expression in characterizing gene regulation. However, the analysis of DAST is challenging because only a small fraction of RNA-seq reads is informative for isoforms. Several methods have been developed to detect exon-based and gene-based DAST, but they suffer from power loss for genes with many isoforms.
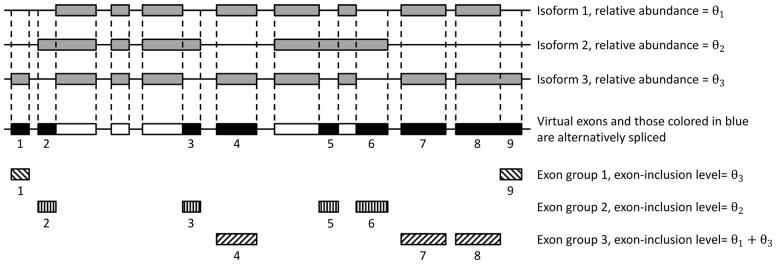
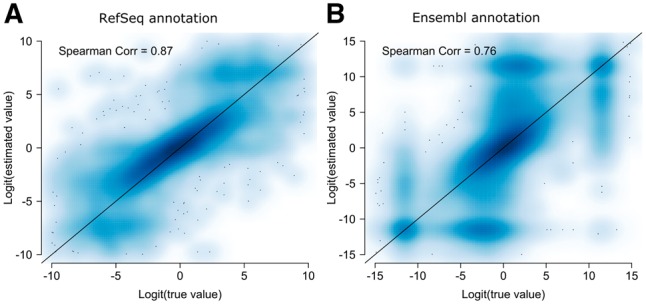
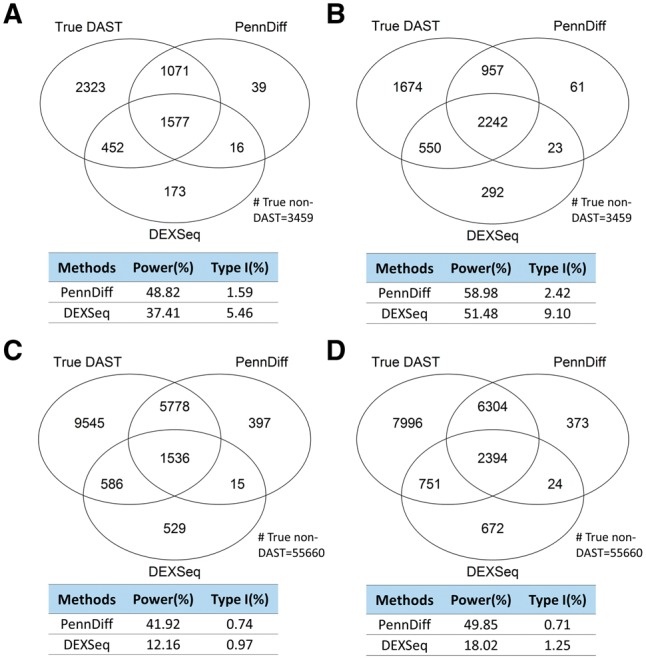
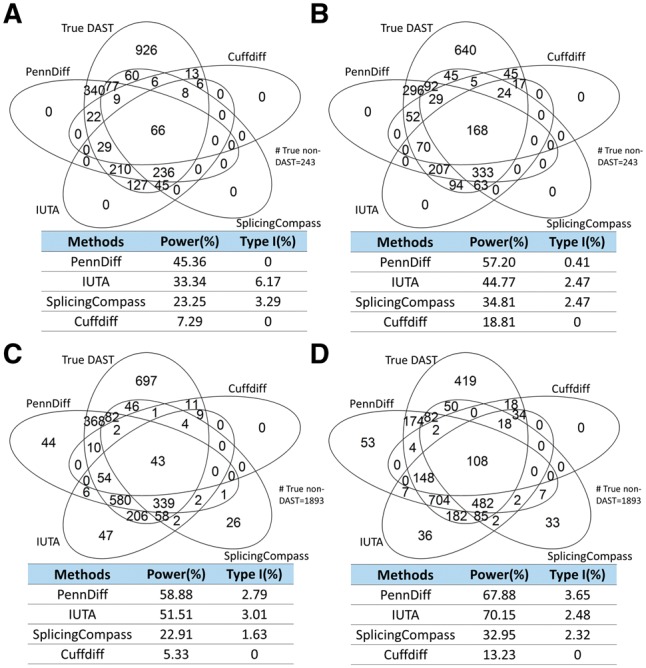
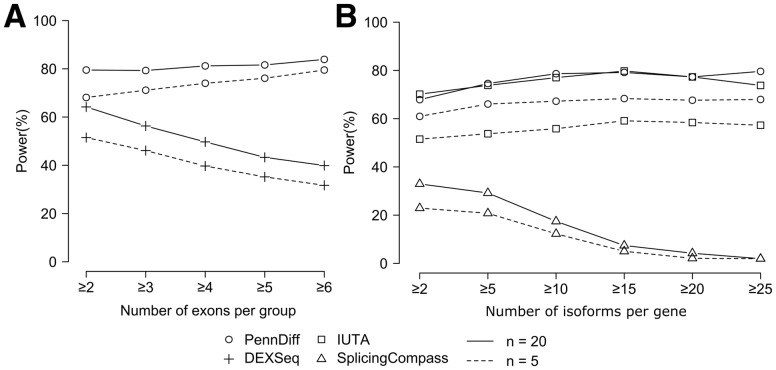
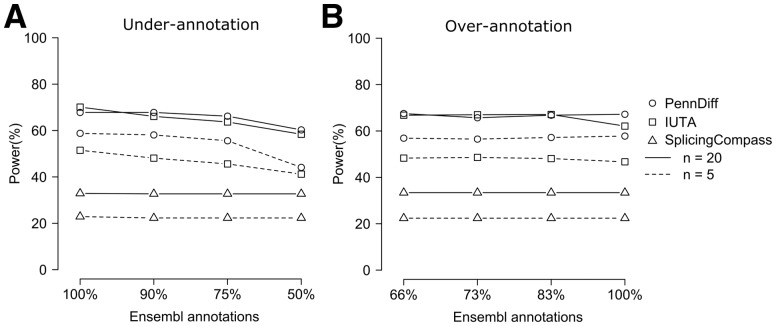
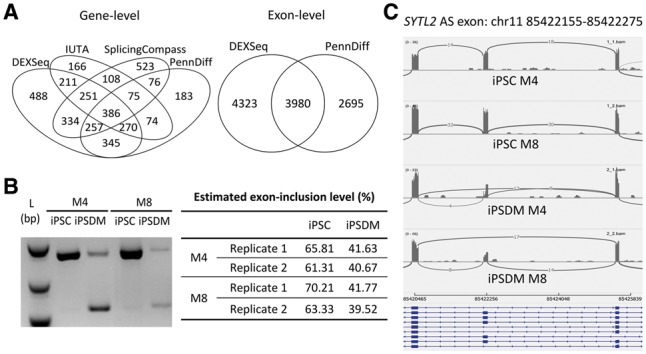
Results: We present PennDiff, a novel statistical method that makes use of information on gene structures and pre-estimated isoform relative abundances, to detect DAST from RNA-seq data. PennDiff has several advantages. First, grouping exons avoids multiple testing for 'exons' originated from the same isoform(s). Second, it utilizes all available reads in exon-inclusion level estimation, which is different from methods that only use junction reads. Third, collapsing isoforms sharing the same alternative exons reduces the impact of isoform expression estimation uncertainty. PennDiff is able to detect DAST at both exon and gene levels, thus offering more flexibility than existing methods. Simulations and analysis of a real RNA-seq dataset indicate that PennDiff has well-controlled type I error rate, and is more powerful than existing methods including DEXSeq, rMATS, Cuffdiff, IUTA and SplicingCompass. As the popularity of RNA-seq continues to grow, we expect PennDiff to be useful for diverse transcriptomics studies.
Availability and implementation: PennDiff source code and user guide is freely available for download at https://github.com/tigerhu15/PennDiff.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
References
-
- Aschoff M. et al. (2013) SplicingCompass: differential splicing detection using RNA-seq data. Bioinformatics, 29, 1141–1148. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
