

Hepatic loss of Lissencephaly 1 (Lis1) induces fatty liver and accelerates liver tumorigenesis in mice
- PMID: 29475944
- PMCID: PMC5892582
- DOI: 10.1074/jbc.RA117.001474
Hepatic loss of Lissencephaly 1 (Lis1) induces fatty liver and accelerates liver tumorigenesis in mice
Abstract
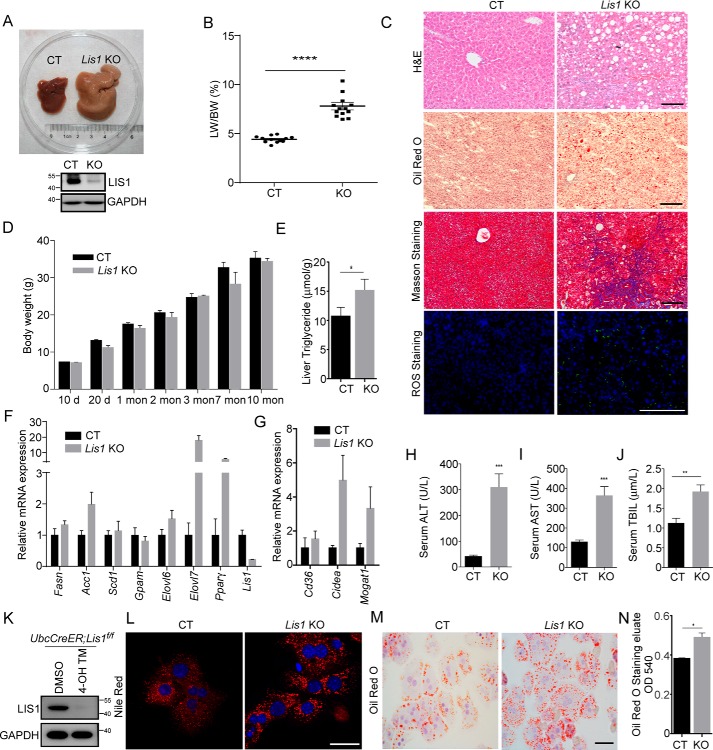
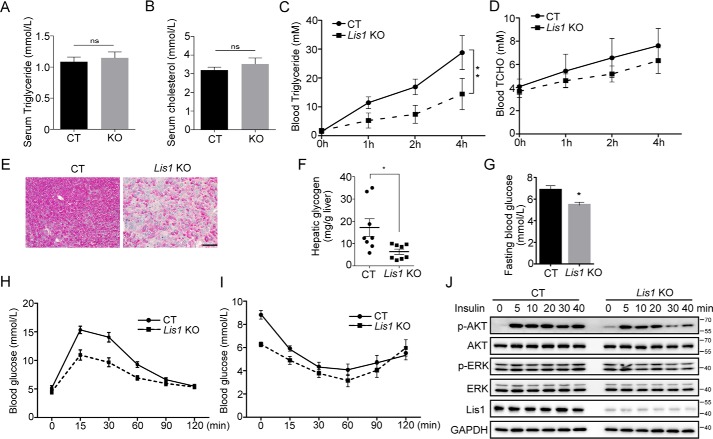
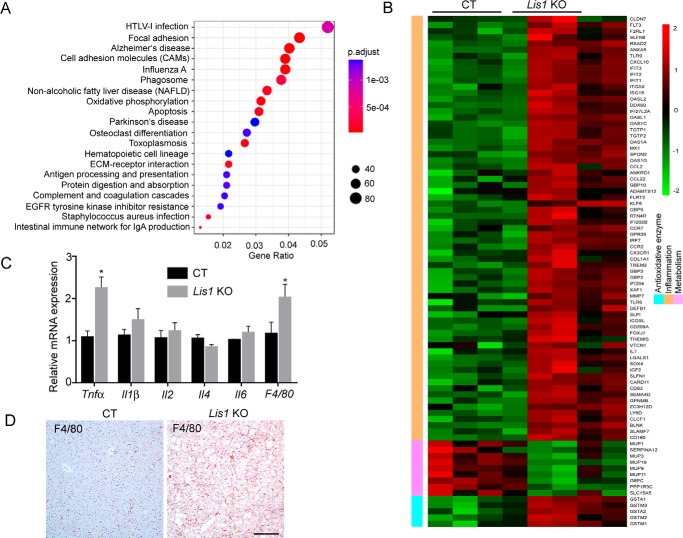
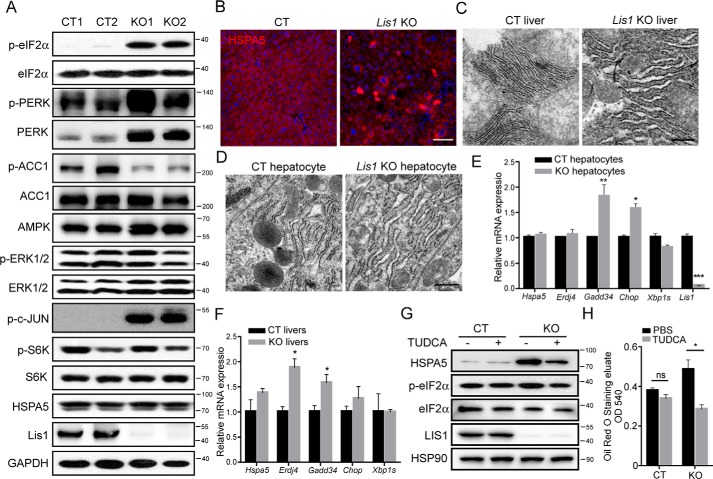
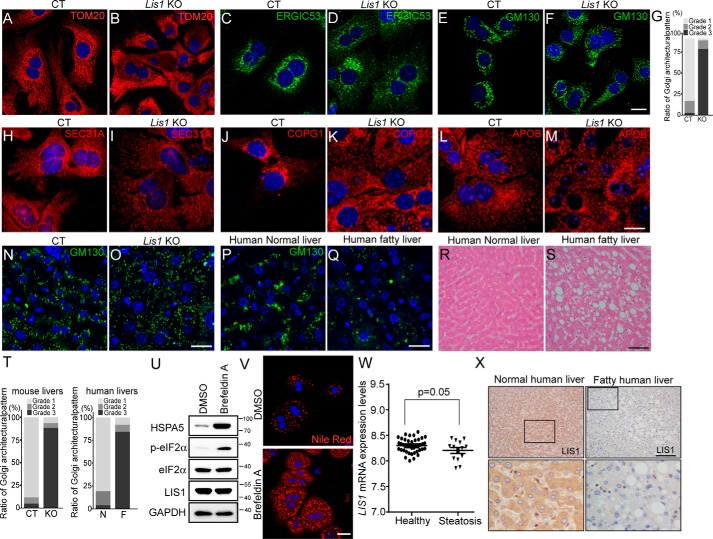
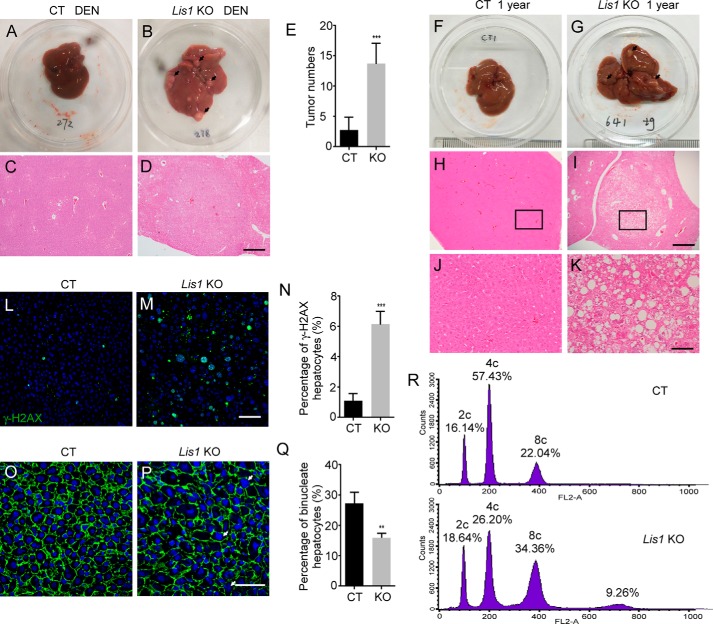
The liver is a major organ in lipid metabolism, and its malfunction leads to various diseases. Nonalcoholic fatty liver disease, the most common chronic liver disorder in developed countries, is characterized by the abnormal retention of excess lipid within hepatocytes and predisposes individuals to liver cancer. We previously reported that the levels of Lissencephaly 1 (LIS1, also known as PAFAH1B1) are down-regulated in human hepatocellular carcinoma. Following up on this observation, we found that genetic deletion of Lis1 in the mouse liver increases lipid accumulation and inflammation in this organ. Further analysis revealed that loss of Lis1 triggers endoplasmic reticulum (ER) stress and reduces triglyceride secretion. Attenuation of ER stress by addition of tauroursodeoxycholic acid (TUDCA) diminished lipid accumulation in the Lis1-deficient hepatocytes. Moreover, the Golgi stacks were disorganized in Lis1-deficient liver cells. Of note, the Lis1 liver-knockout mice exhibited increased hepatocyte ploidy and accelerated development of liver cancer after exposure to the liver carcinogen diethylnitrosamine (DEN). Taken together, these findings suggest that reduced Lis1 levels can spur the development of liver diseases from steatosis to liver cancer and provide a useful model for delineating the molecular pathways that lead to these diseases.
Keywords: Golgi; Lis1; endoplasmic reticulum stress (ER stress); hepatocyte; liver cancer; liver metabolism; mouse genetics; nonalcoholic fatty liver disease; ploidy; steatosis.
© 2018 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
