

Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans
- PMID: 29477724
- PMCID: PMC6148345
- DOI: 10.1016/j.jaci.2018.01.039
Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans
Abstract
Background: The genetic cause of primary immunodeficiency disease (PID) carries prognostic information.
Objective: We conducted a whole-genome sequencing study assessing a large proportion of the NIHR BioResource-Rare Diseases cohort.
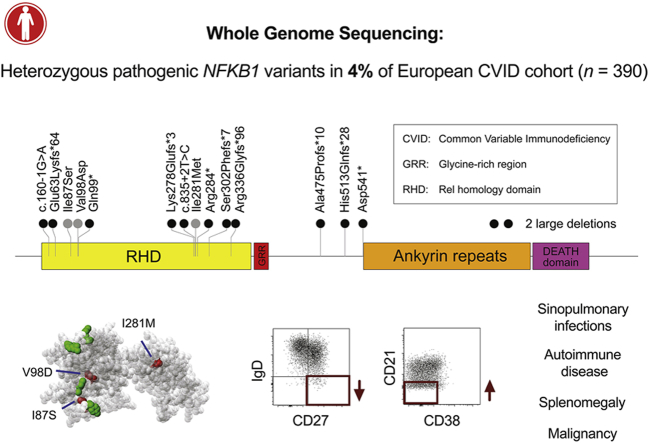
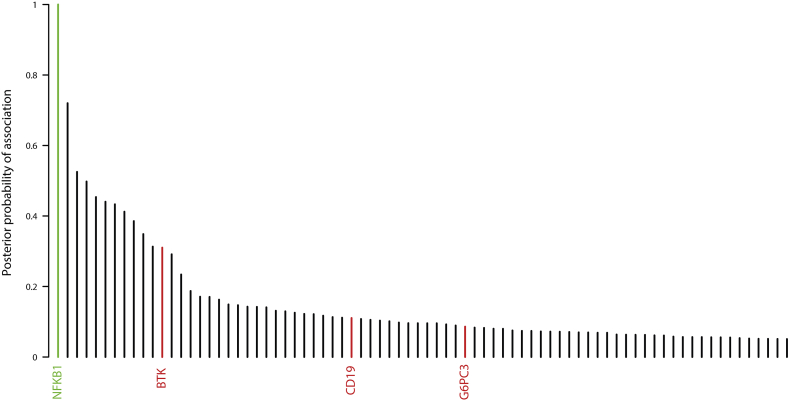
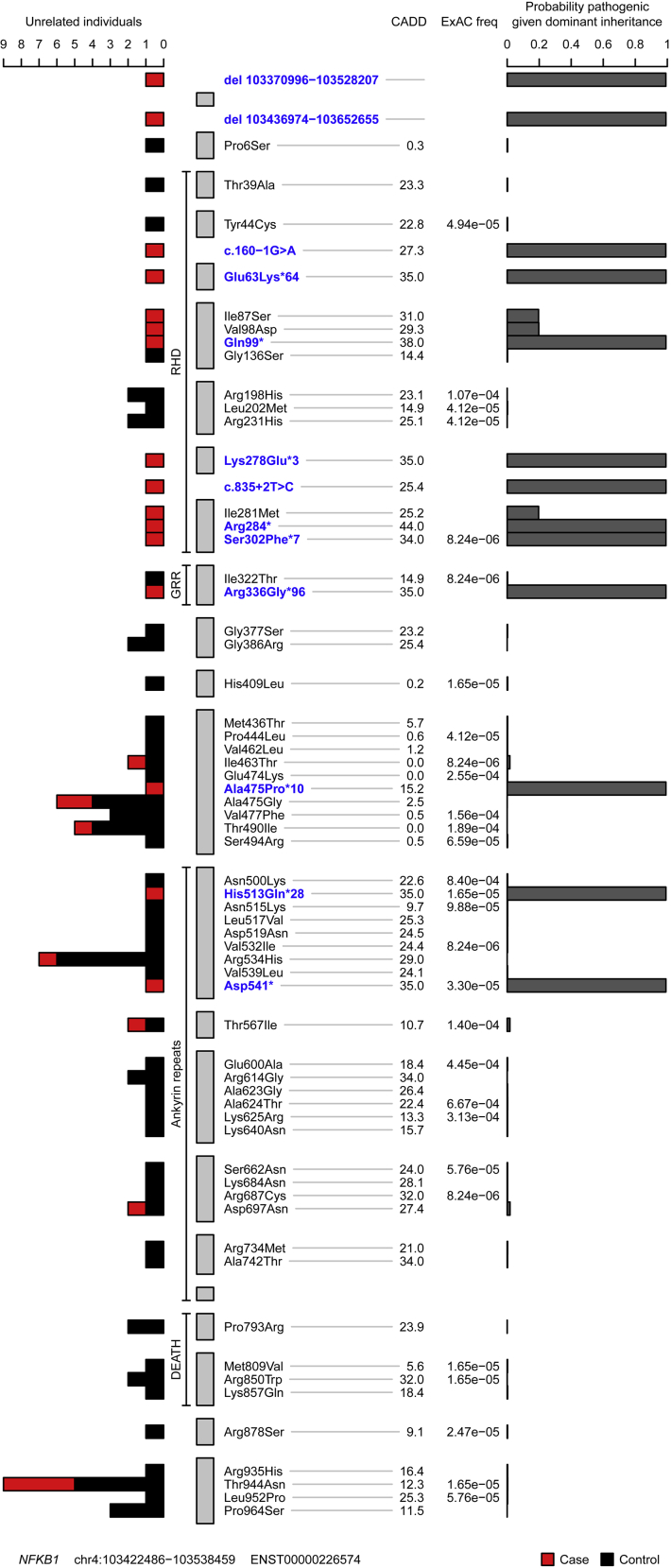
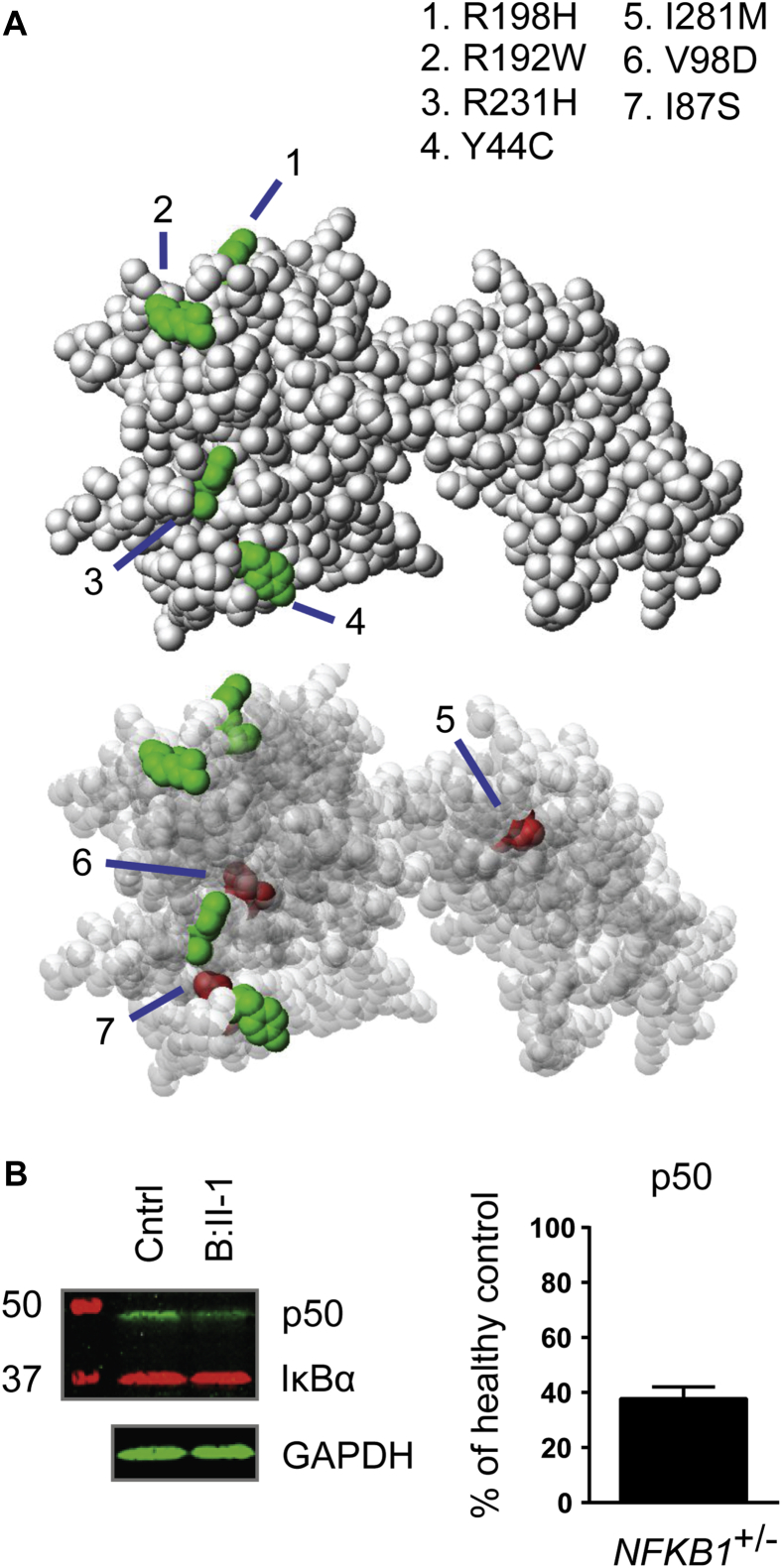
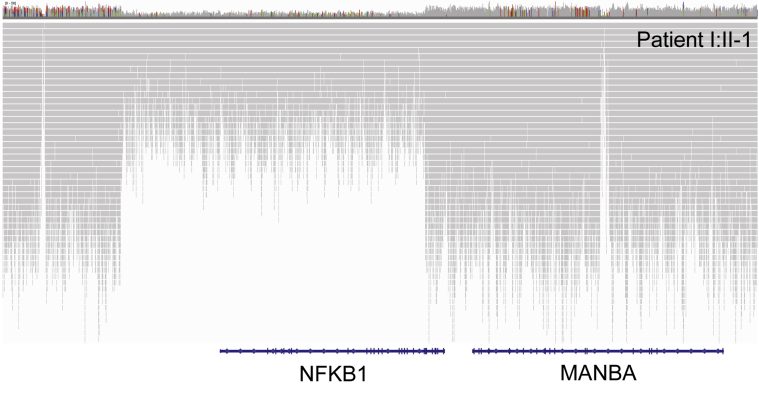
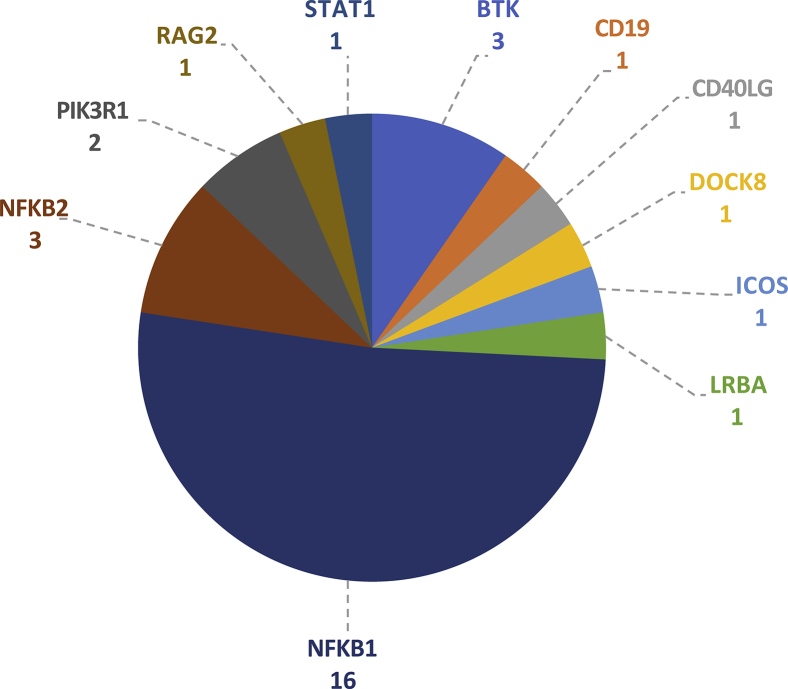
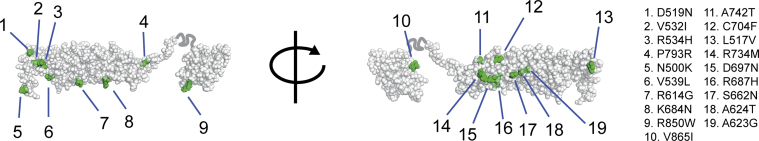
Methods: In the predominantly European study population of principally sporadic unrelated PID cases (n = 846), a novel Bayesian method identified nuclear factor κB subunit 1 (NFKB1) as one of the genes most strongly associated with PID, and the association was explained by 16 novel heterozygous truncating, missense, and gene deletion variants. This accounted for 4% of common variable immunodeficiency (CVID) cases (n = 390) in the cohort. Amino acid substitutions predicted to be pathogenic were assessed by means of analysis of structural protein data. Immunophenotyping, immunoblotting, and ex vivo stimulation of lymphocytes determined the functional effects of these variants. Detailed clinical and pedigree information was collected for genotype-phenotype cosegregation analyses.
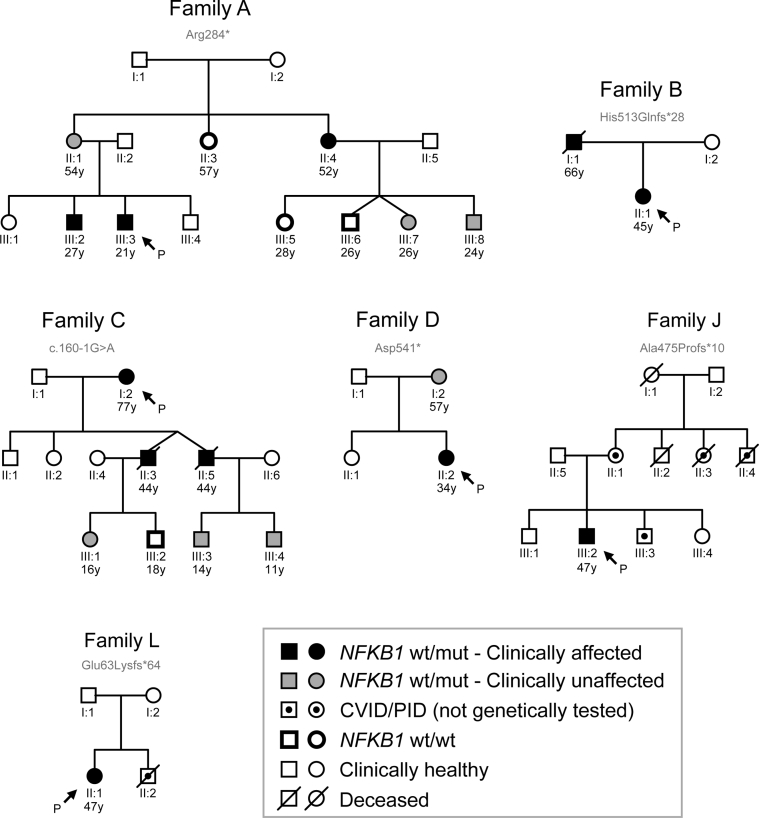
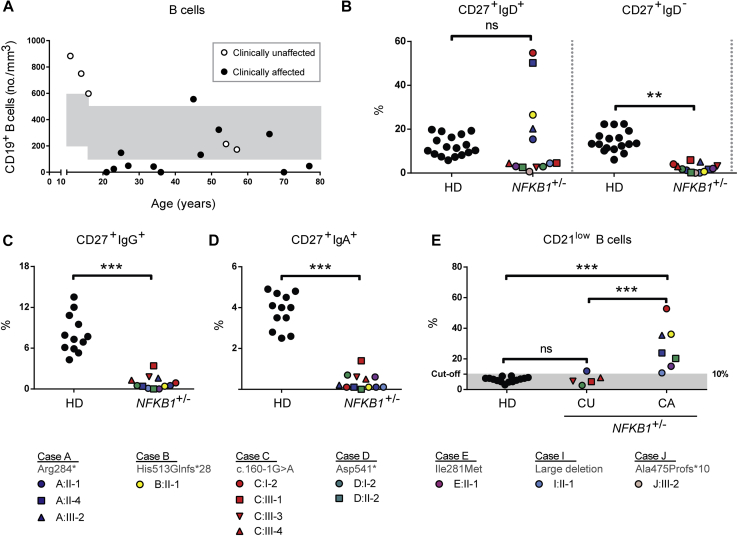
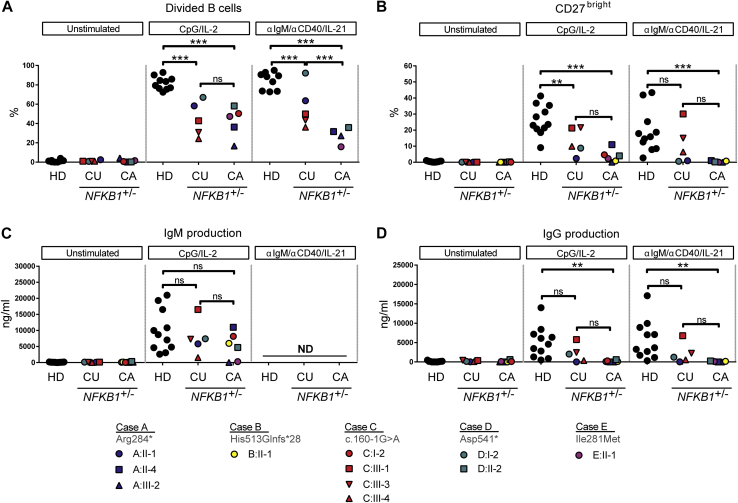
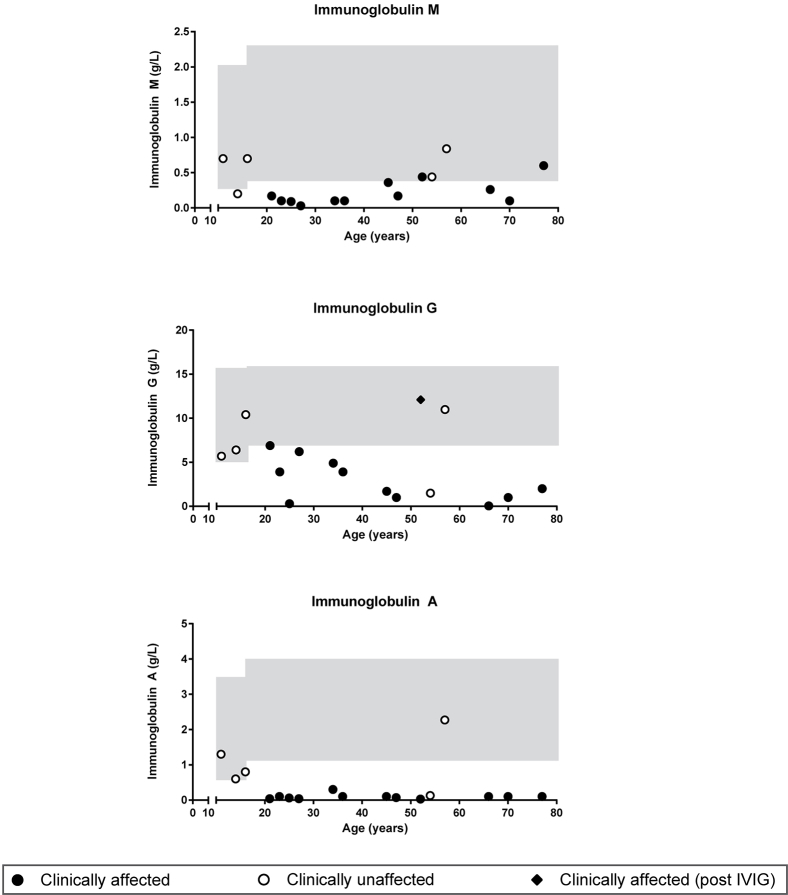
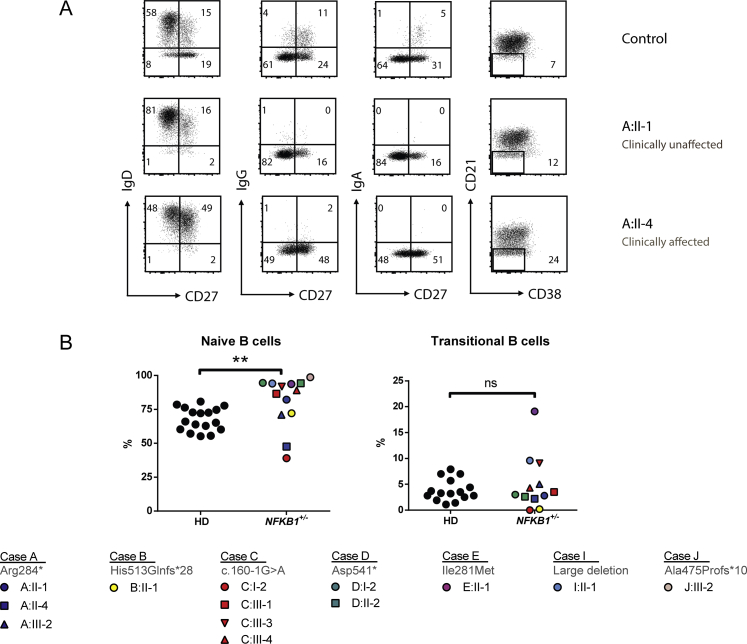
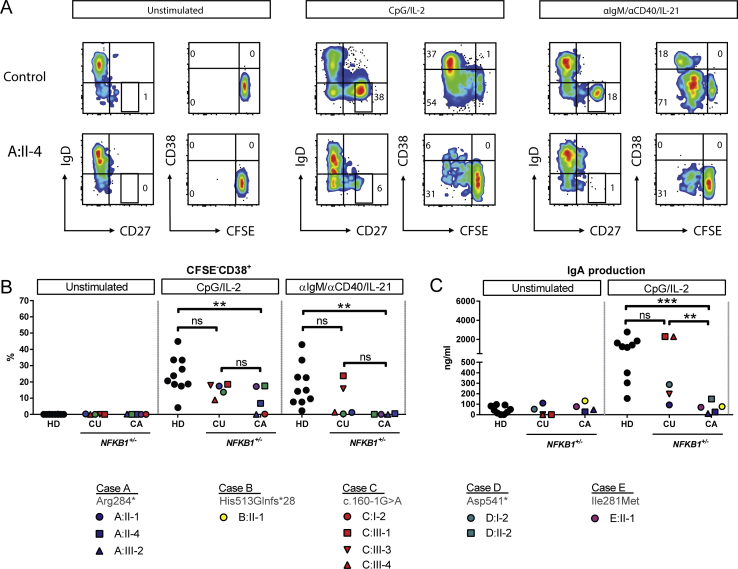
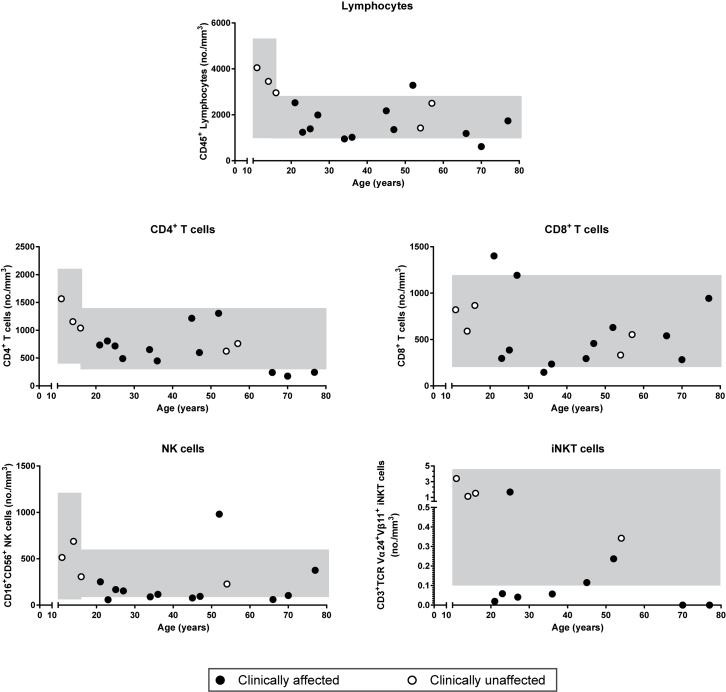
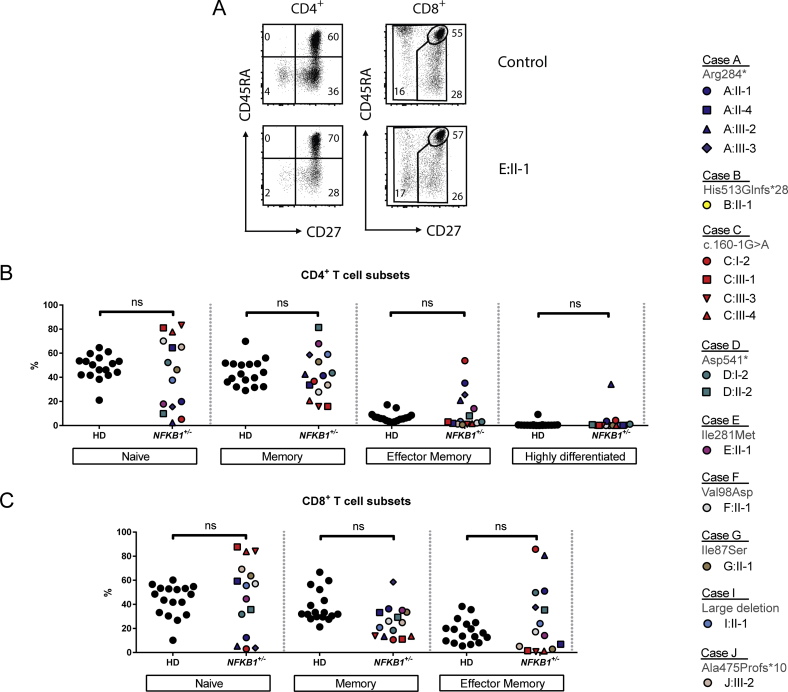
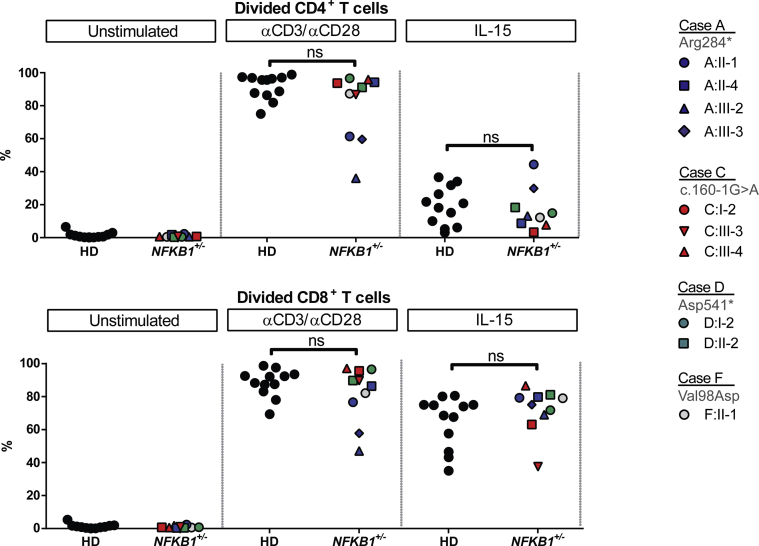
Results: Both sporadic and familial cases demonstrated evidence of the noninfective complications of CVID, including massive lymphadenopathy (24%), unexplained splenomegaly (48%), and autoimmune disease (48%), features prior studies correlated with worse clinical prognosis. Although partial penetrance of clinical symptoms was noted in certain pedigrees, all carriers have a deficiency in B-lymphocyte differentiation. Detailed assessment of B-lymphocyte numbers, phenotype, and function identifies the presence of an increased CD21low B-cell population. Combined with identification of the disease-causing variant, this distinguishes between healthy subjects, asymptomatic carriers, and clinically affected cases.
Conclusion: We show that heterozygous loss-of-function variants in NFKB1 are the most common known monogenic cause of CVID, which results in a temporally progressive defect in the formation of immunoglobulin-producing B cells.
Keywords: B cells; common variable immunodeficiency; nuclear factor κB1.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Figures

Comment in
-
Nuclear factor κB mutations in human subjects: The devil is in the details.J Allergy Clin Immunol. 2018 Oct;142(4):1062-1065. doi: 10.1016/j.jaci.2018.06.050. Epub 2018 Aug 28. J Allergy Clin Immunol. 2018. PMID: 30165054 No abstract available.
References
-
- Primary immunodeficiency diseases. Report of a WHO scientific group. Clin Exp Immunol. 1997;109(Suppl 1):1–28. - PubMed
-
- Oksenhendler E., Gerard L., Fieschi C., Malphettes M., Mouillot G., Jaussaud R. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. 2008;46:1547–1554. - PubMed
-
- Cunningham-Rundles C., Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34–48. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
