

Carcinogenesis and Reactive Oxygen Species Signaling: Interaction of the NADPH Oxidase NOX1-5 and Superoxide Dismutase 1-3 Signal Transduction Pathways
- PMID: 29478325
- PMCID: PMC6393772
- DOI: 10.1089/ars.2017.7268
Carcinogenesis and Reactive Oxygen Species Signaling: Interaction of the NADPH Oxidase NOX1-5 and Superoxide Dismutase 1-3 Signal Transduction Pathways
Abstract
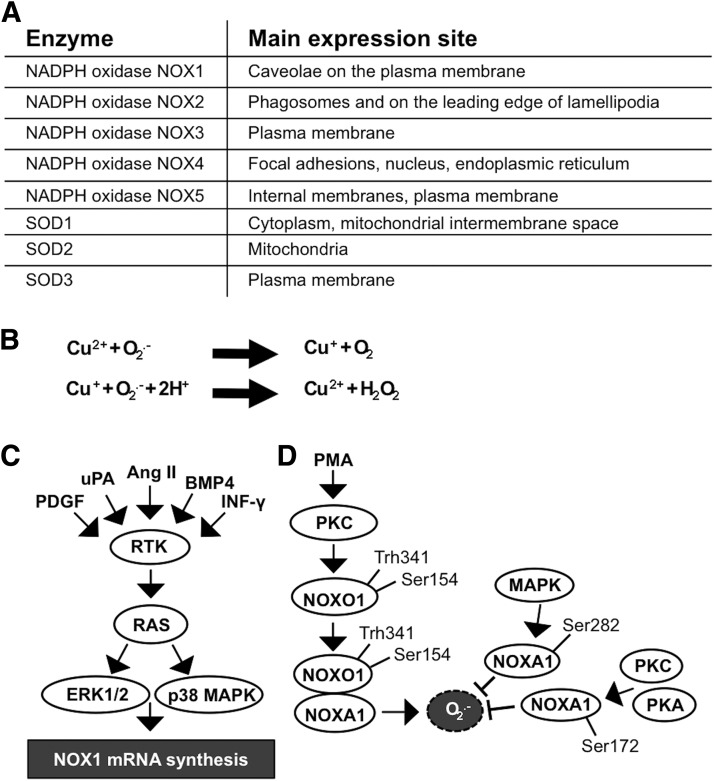
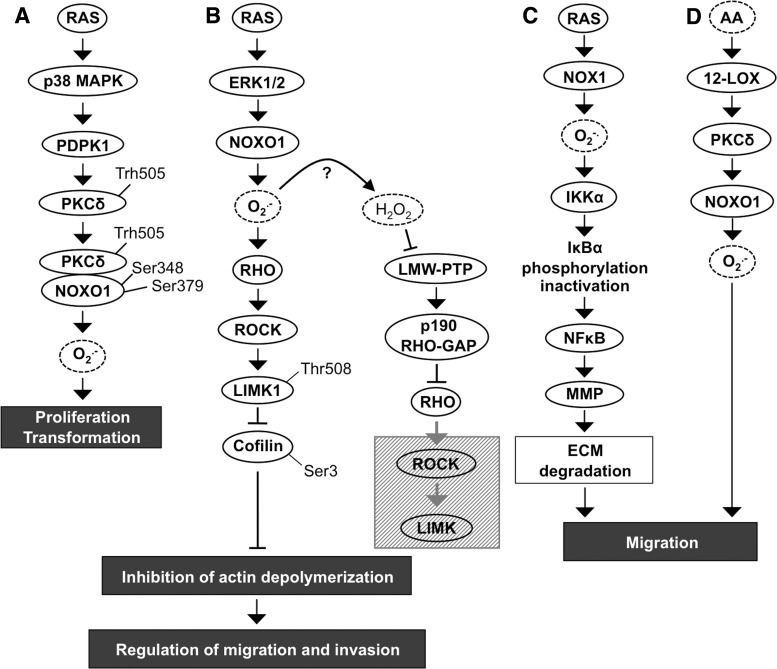
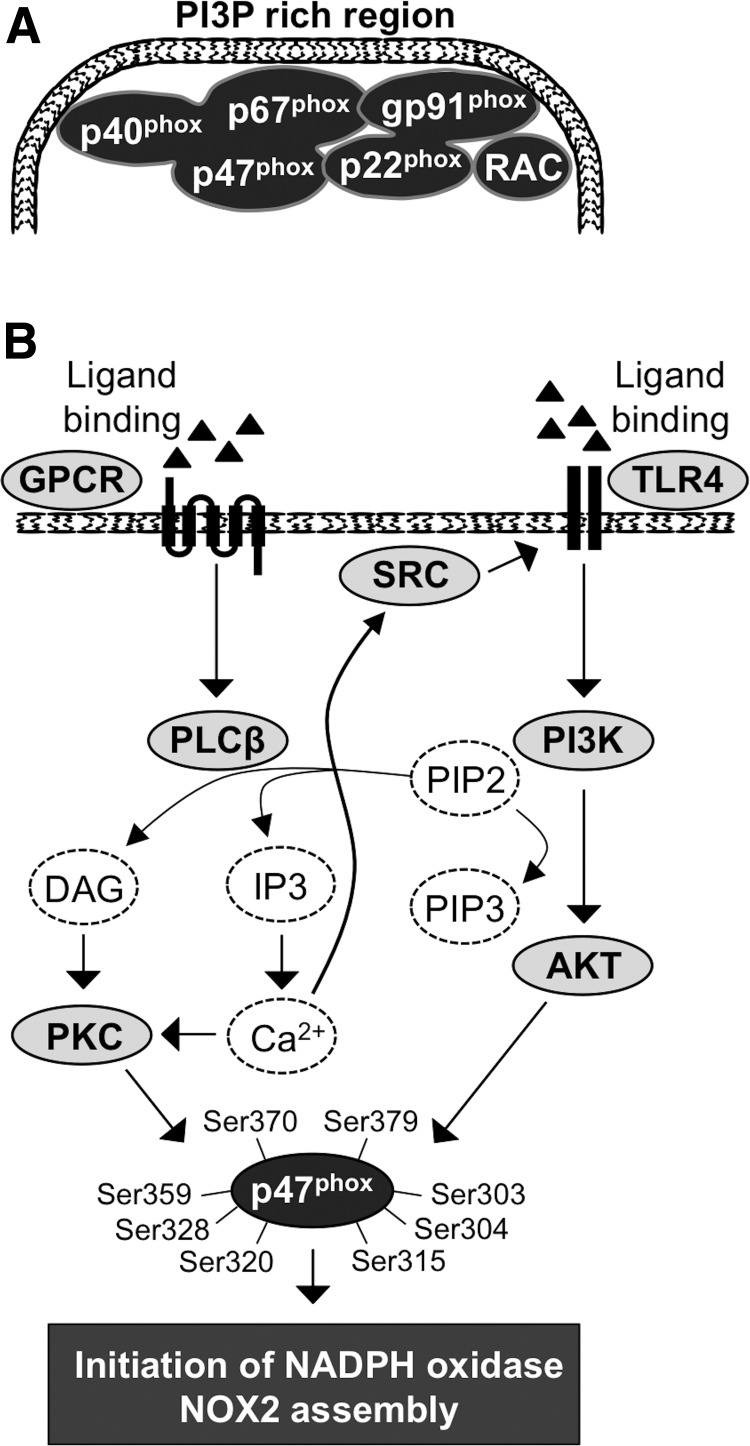
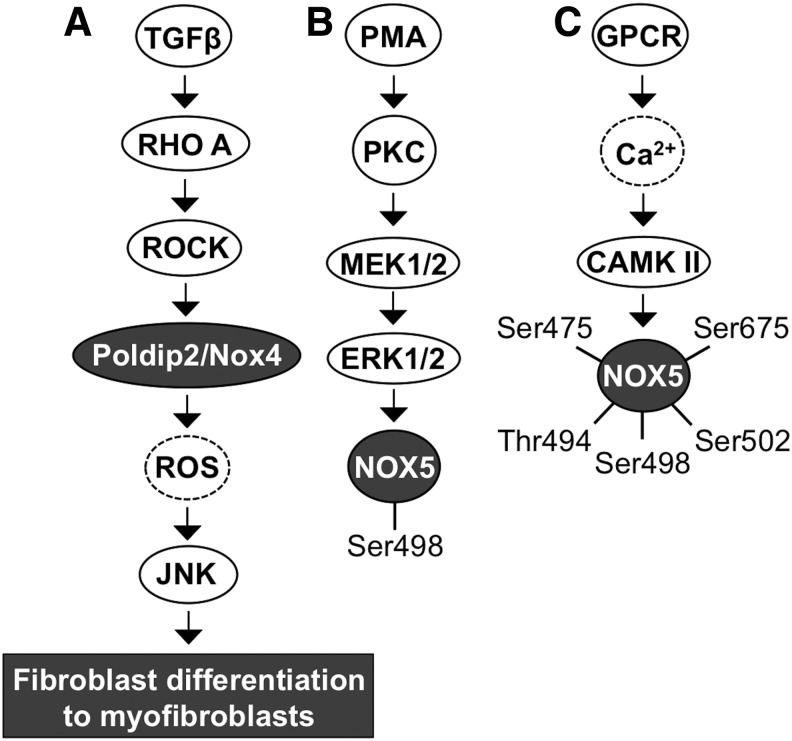
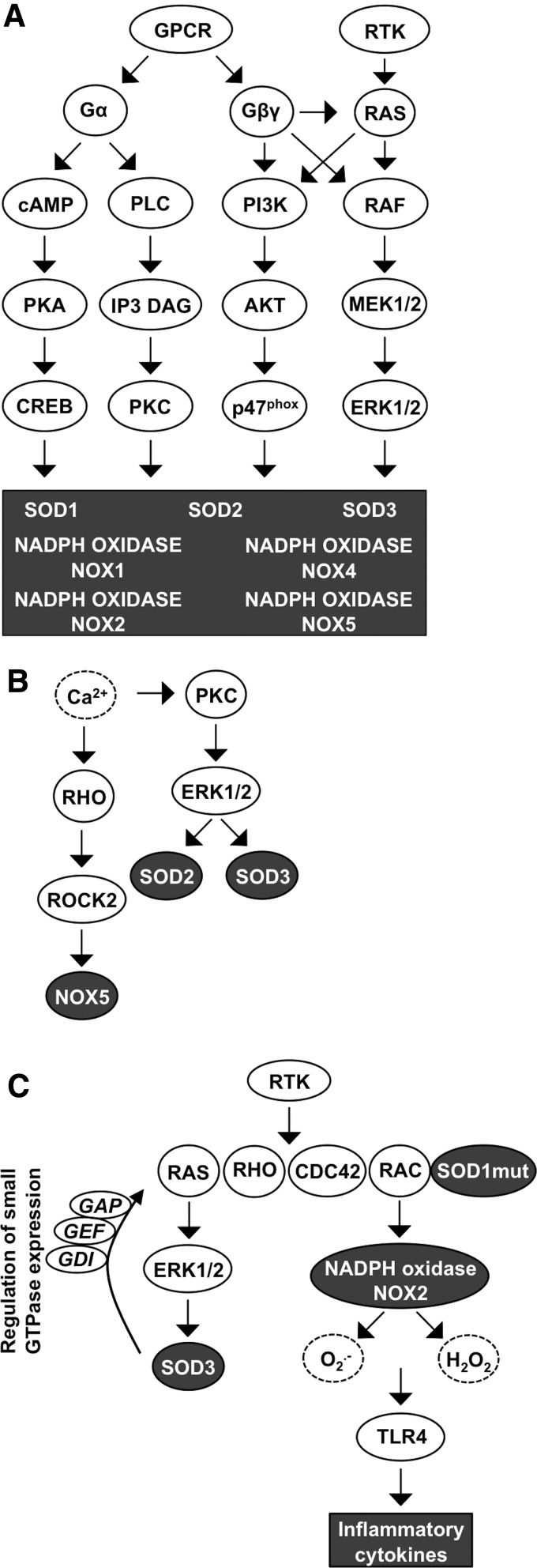
Significance: Reduction/oxidation (redox) balance could be defined as an even distribution of reduction and oxidation complementary processes and their reaction end products. There is a consensus that aberrant levels of reactive oxygen species (ROS), commonly observed in cancer, stimulate primary cell immortalization and progression of carcinogenesis. However, the mechanism how different ROS regulate redox balance is not completely understood. Recent Advances: In the current review, we have summarized the main signaling cascades inducing NADPH oxidase NOX1-5 and superoxide dismutase (SOD) 1-3 expression and their connection to cell proliferation, immortalization, transformation, and CD34+ cell differentiation in thyroid, colon, lung, breast, and hematological cancers.
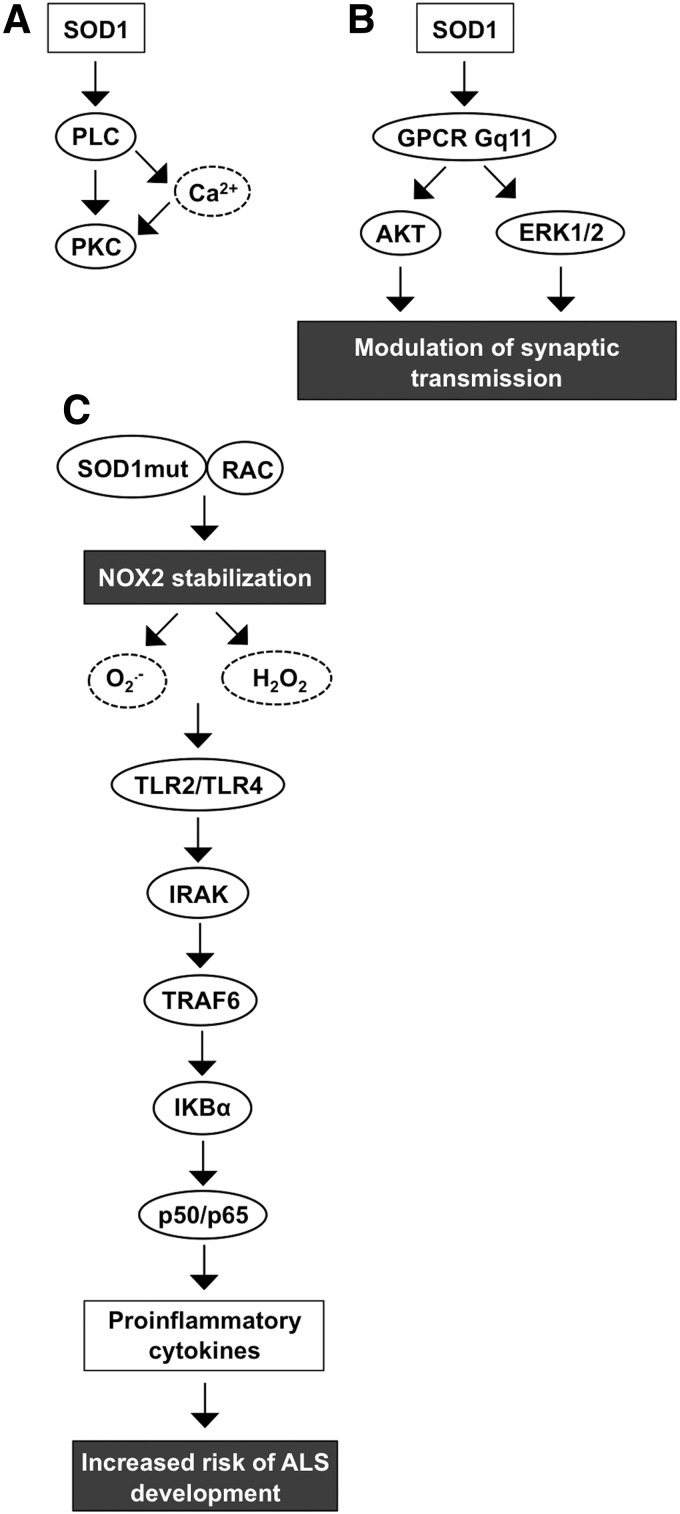
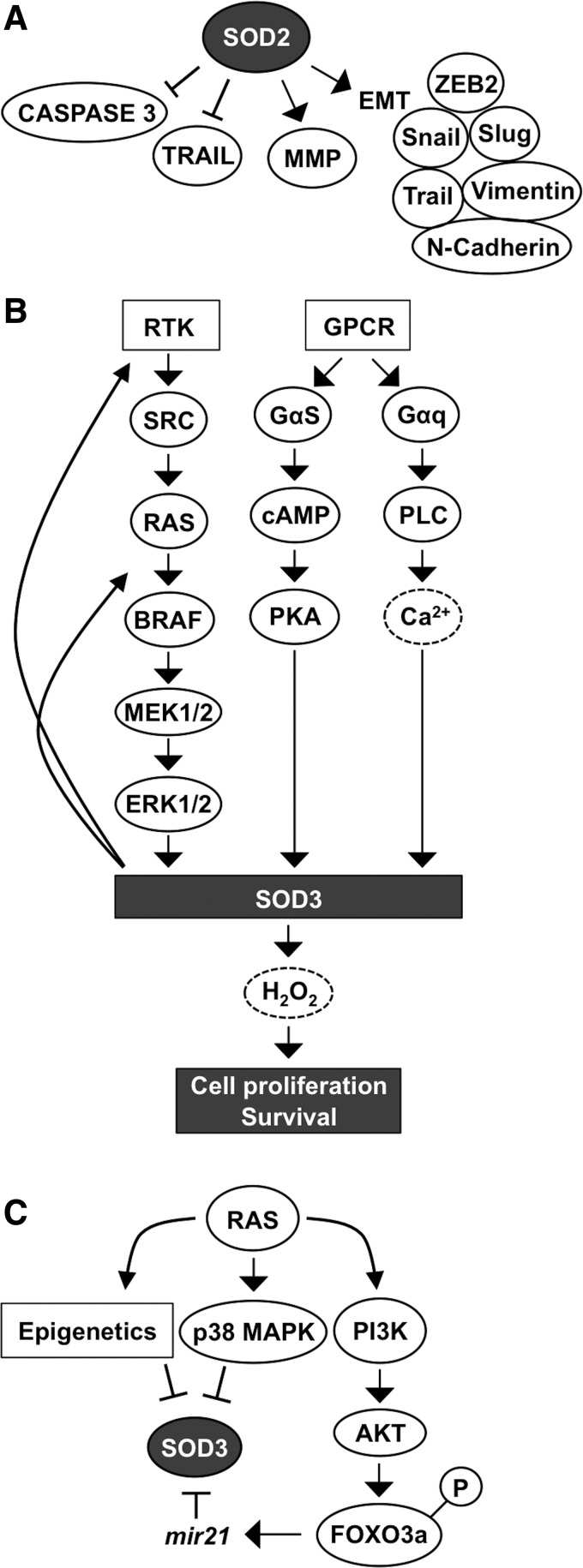
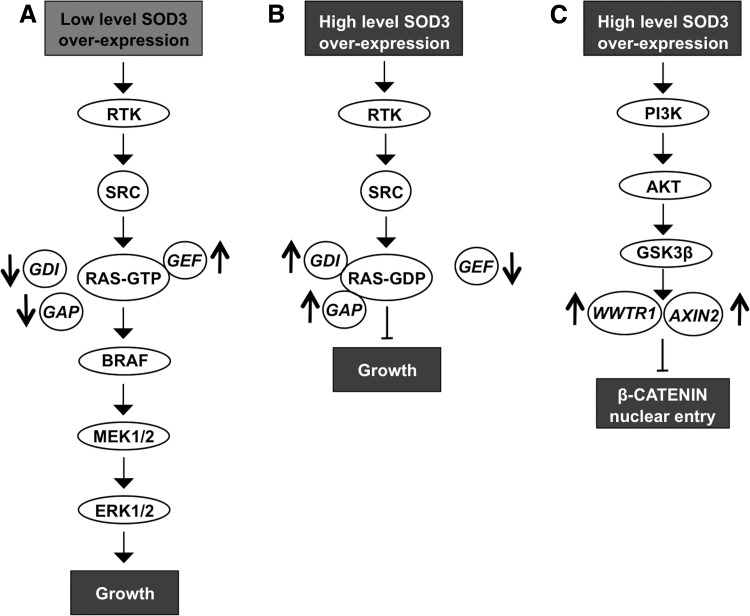
Critical issues: Interestingly, many of the signaling pathways activating redox enzymes or mediating the effect of ROS are common, such as pathways initiated from G protein-coupled receptors and tyrosine kinase receptors involving protein kinase A, phospholipase C, calcium, and small GTPase signaling molecules.
Future directions: The clarification of interaction of signal transduction pathways could explain how cells regulate redox balance and may even provide means to inhibit the accumulation of harmful levels of ROS in human pathologies.
Keywords: G protein-coupled receptor; NADPH oxidase NOX; redox signaling; superoxide dismutase; tyrosine kinase receptor.
Figures
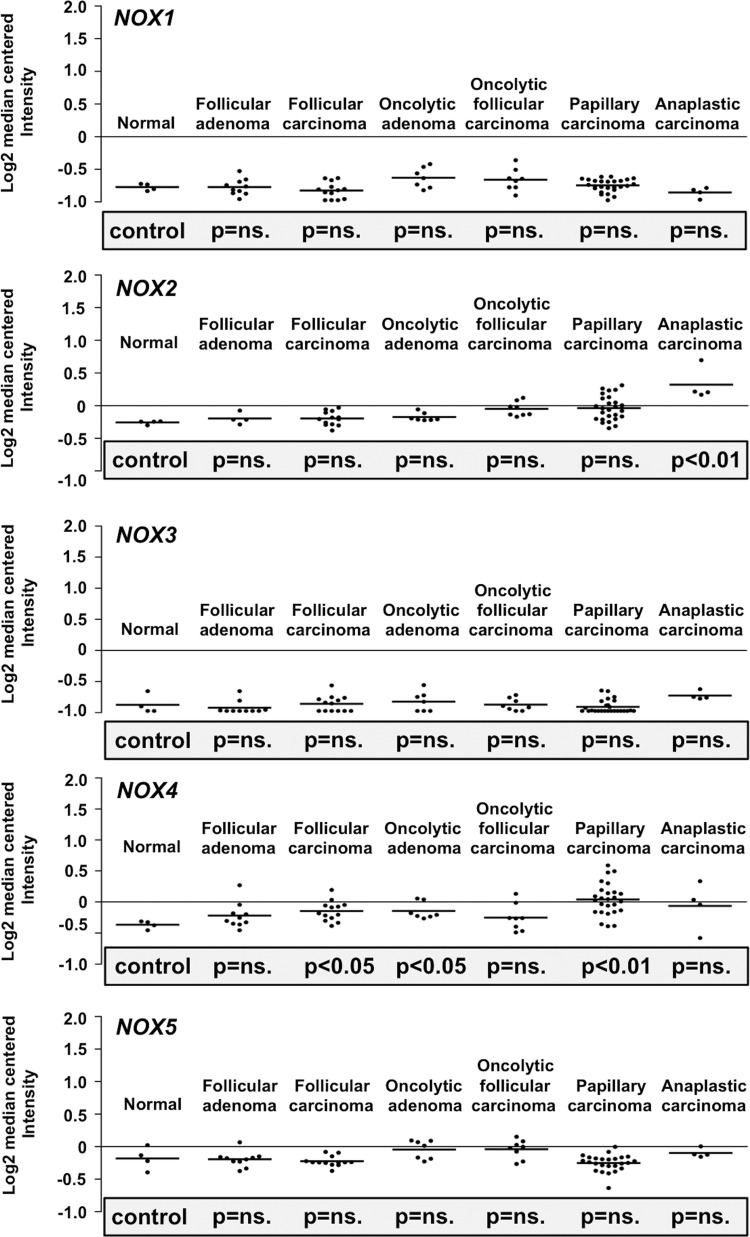

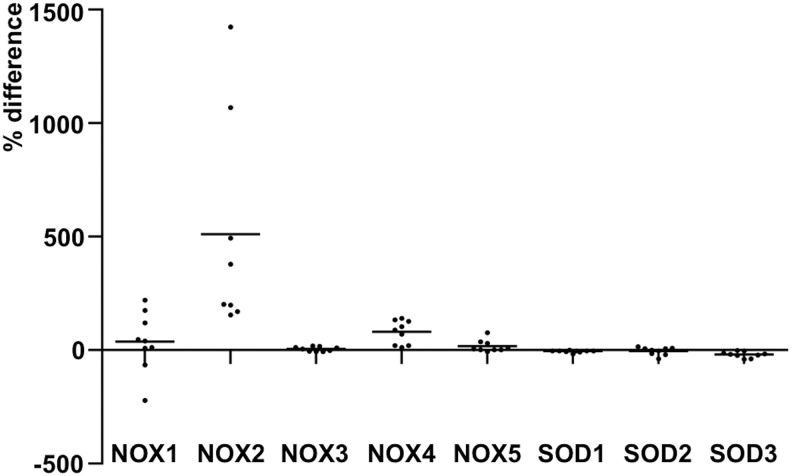
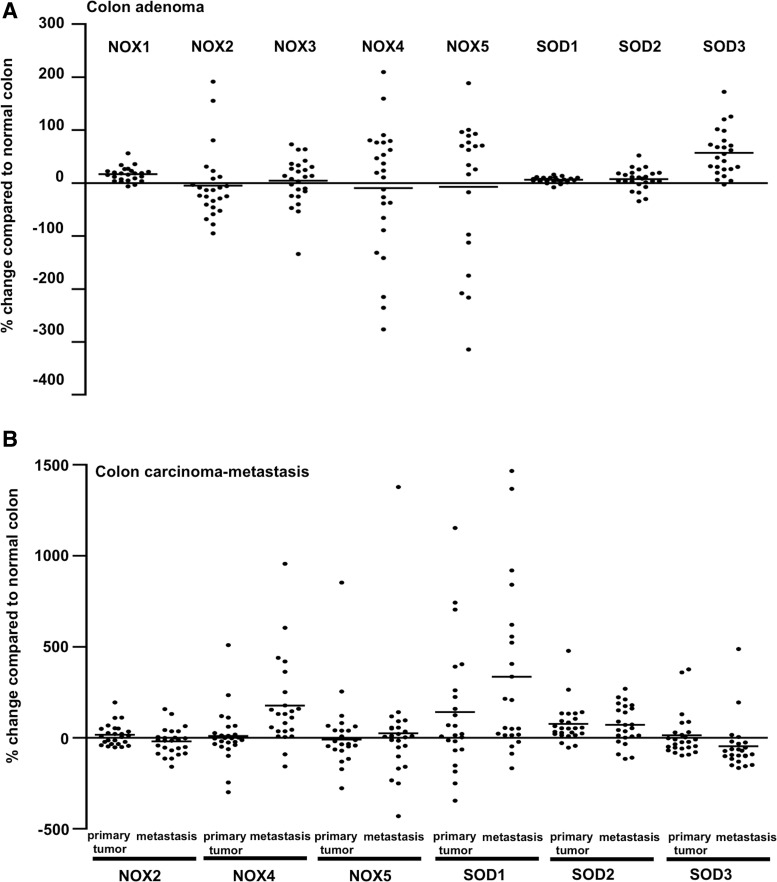
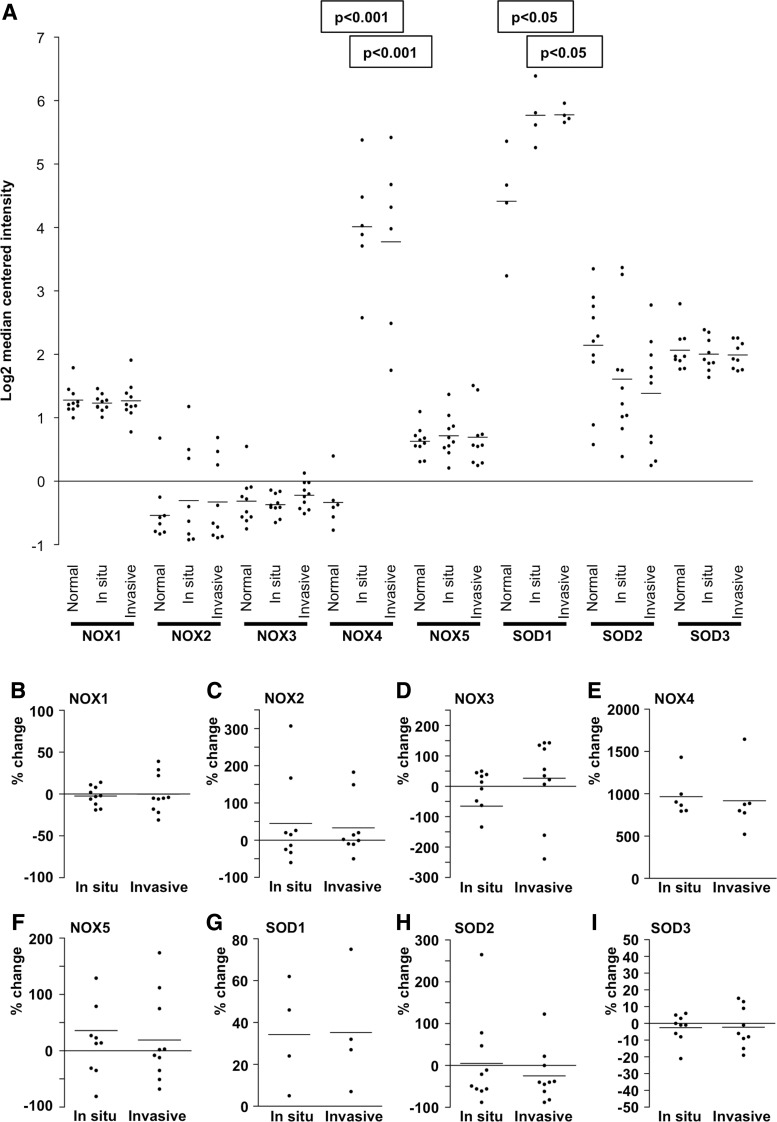
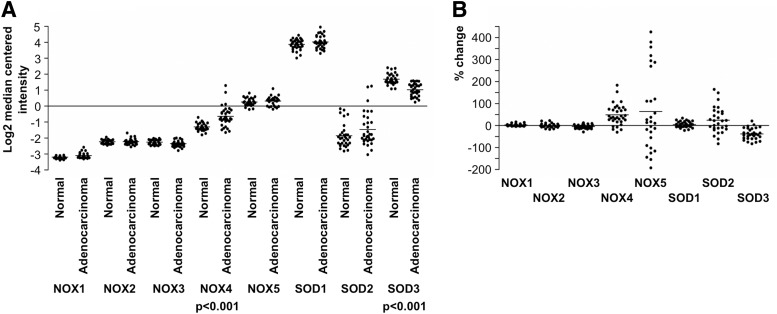
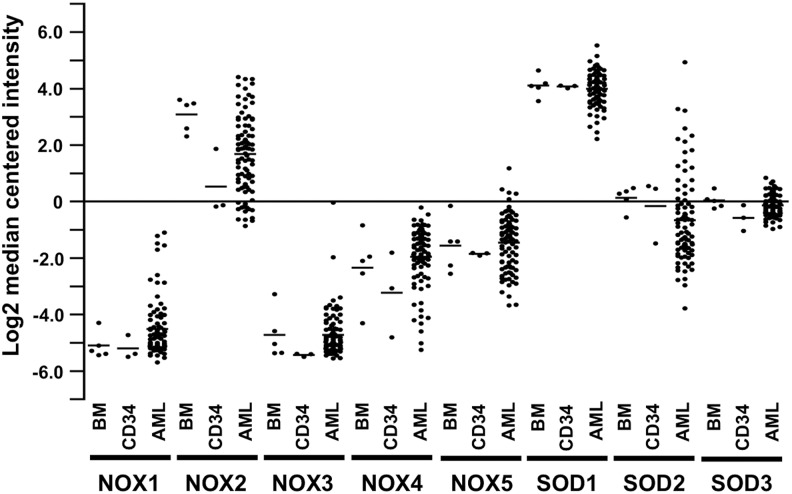
References
-
- Abdel-Mageed AS, Senagore AJ, Pietryga DW, Connors RH, Giambernardi TA, Hay RV, and Deng W. Intravenous administration of mesenchymal stem cells genetically modified with extracellular superoxide dismutase improves survival in irradiated mice. Blood 113: 1201–1203, 2009. - PubMed
-
- Abrahamsson T, Brandt U, Marklund SL, and Sjoqvist PO. Vascular bound recombinant extracellular superoxide dismutase type C protects against the detrimental effects of superoxide radicals on endothelium-dependent arterial relaxation. Circ Res 70: 264–271, 1992. - PubMed
-
- Adachi T, Toishi T, Takashima E, and Hara H. Infliximab neutralizes the suppressive effect of TNF-alpha on expression of extracellular-superoxide dismutase in vitro. Biol Pharm Bull 29: 2095–2098, 2006. - PubMed
-
- Adachi T, Yamada H, Futenma A, Kato K, and Hirano K. Heparin-induced release of extracellular-superoxide dismutase form (V) to plasma. J Biochem 117: 586–590, 1995. - PubMed
-
- Adachi Y, Shibai Y, Mitsushita J, Shang WH, Hirose K, and Kamata T. Oncogenic Ras upregulates NADPH oxidase 1 gene expression through MEK-ERK-dependent phosphorylation of GATA-6. Oncogene 27: 4921–4932, 2008. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
