

Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications
- PMID: 29478411
- PMCID: PMC6251479
- DOI: 10.1186/s13059-018-1406-4
Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications
Abstract
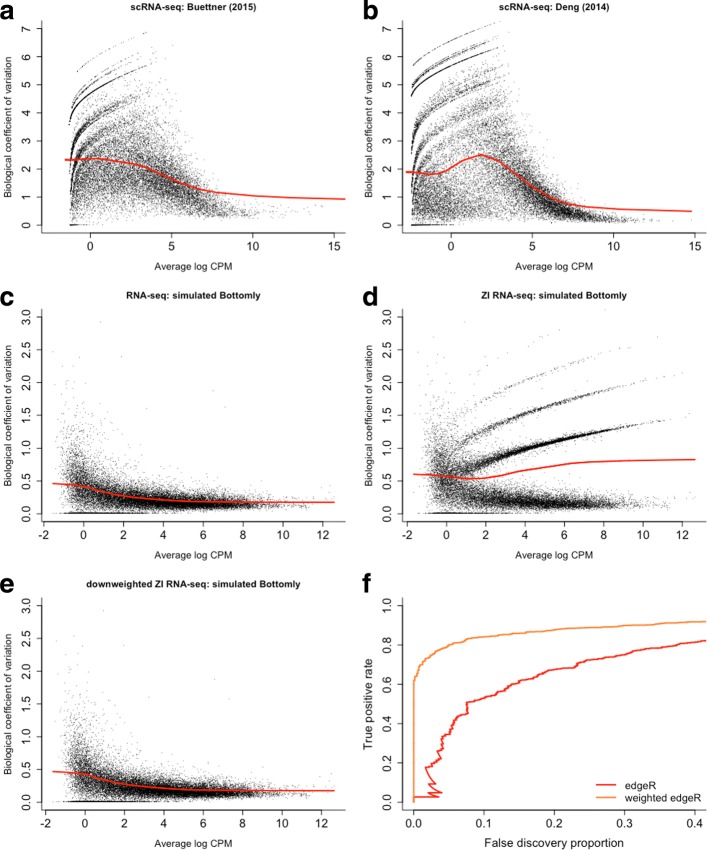
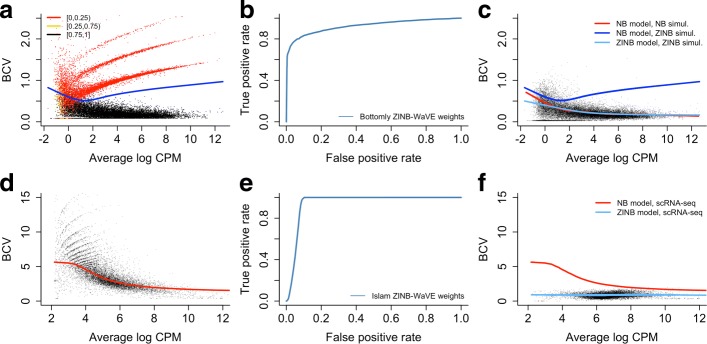
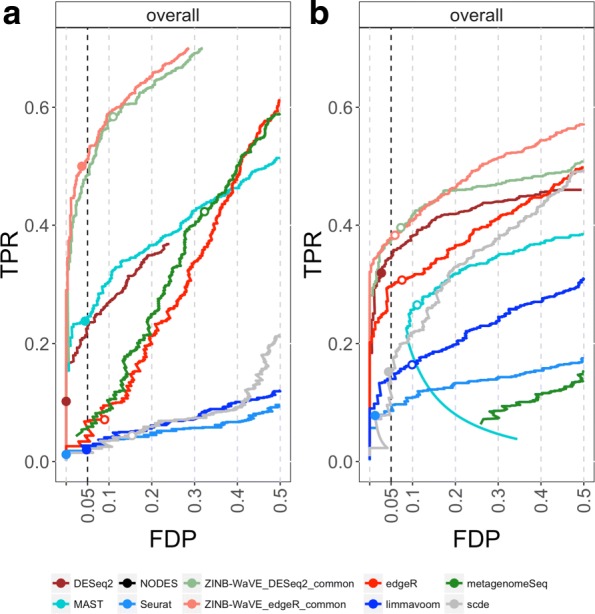
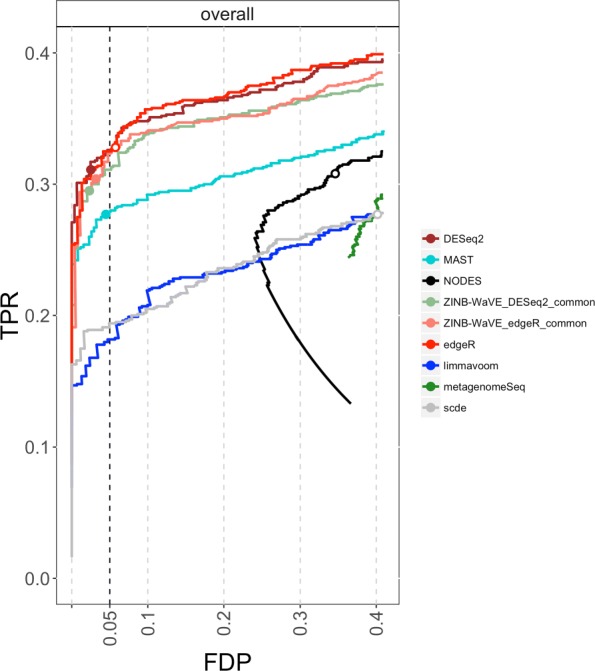
Dropout events in single-cell RNA sequencing (scRNA-seq) cause many transcripts to go undetected and induce an excess of zero read counts, leading to power issues in differential expression (DE) analysis. This has triggered the development of bespoke scRNA-seq DE methods to cope with zero inflation. Recent evaluations, however, have shown that dedicated scRNA-seq tools provide no advantage compared to traditional bulk RNA-seq tools. We introduce a weighting strategy, based on a zero-inflated negative binomial model, that identifies excess zero counts and generates gene- and cell-specific weights to unlock bulk RNA-seq DE pipelines for zero-inflated data, boosting performance for scRNA-seq.
Keywords: Differential expression; Single-cell RNA sequencing; Weights; Zero-inflated negative binomial.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
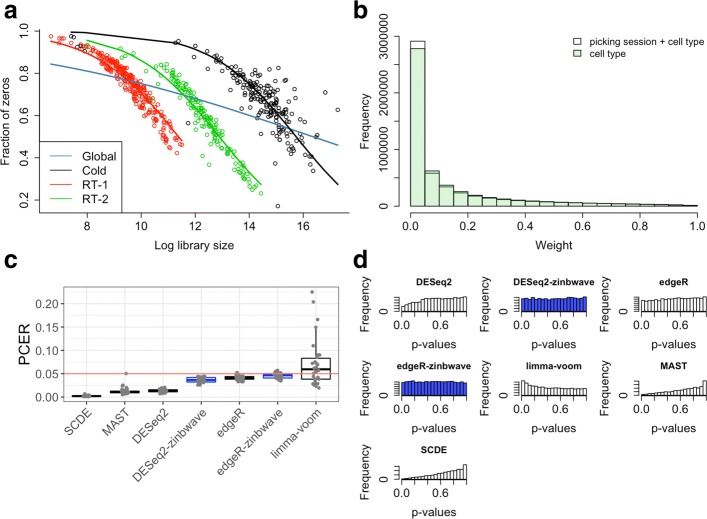
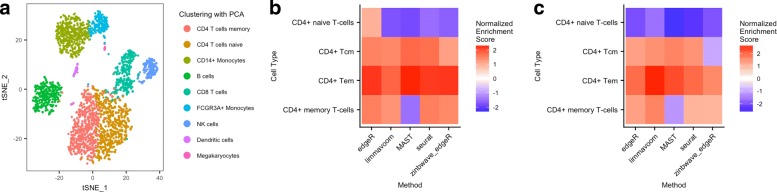
References
-
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15(12):550. http://genomebiology.com/2014/15/12/550. - PMC - PubMed
-
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26(1):139–40. http://www.ncbi.nlm.nih.gov/pubmed/19910308. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC2796818. - PMC - PubMed
-
- Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014; 15(2):R29. http://www.pubmedcentral.nih.gov/articlerender.fcgi%3Fartid=4053721%26to.... - PMC - PubMed
-
- Wang Z, Gerstein M, Snyder M. RNA-seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009; 10(1):57–63. http://www.nature.com/doifinder/10.1038/nrg2484. - DOI - PMC - PubMed
-
- Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016; 17(6):333–51. http://www.nature.com/doifinder/10.1038/nrg.2016.49. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
