

Hereditary Neuropathies
- PMID: 29478438
- PMCID: PMC5832893
- DOI: 10.3238/arztebl.2018.0091
Hereditary Neuropathies
Abstract
Background: Hereditary peripheral neuropathies constitute a large group of genetic diseases, with an overall prevalence of 1:2500. In recent years, the use of so-called next-generation sequencing (NGS) has led to the identification of many previously unknown involved genes and genetic defects that cause neuropathy. In this article, we review the procedures and utility of genetic evaluation for hereditary neurop - athies, while also considering the implications of the fact that causally directed treatment of these disorders is generally unavailable.
Methods: This review is based on pertinent publications retrieved by a PubMed search employing the search terms "hereditary neuropathy," "Charcot-Marie-Tooth disease," "hereditary sensory neuropathy," and "hereditary motor neuropathy."
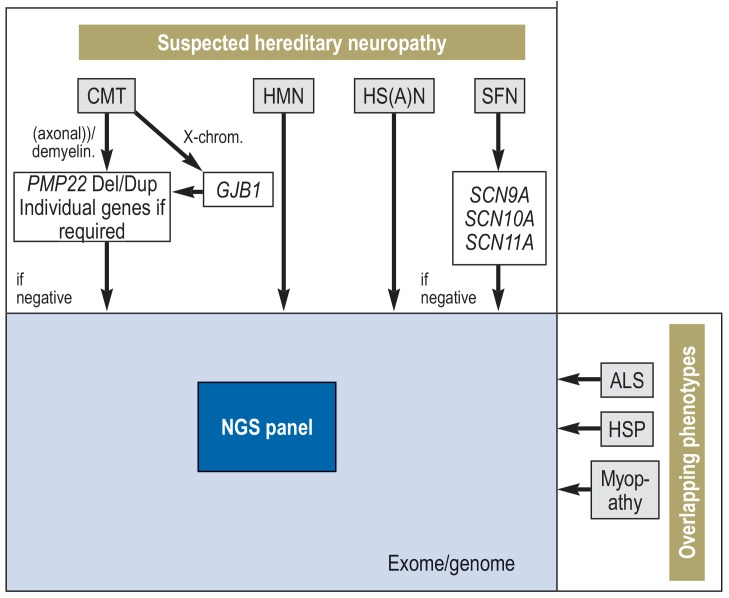
Results: With rare exceptions, the diagnostic evaluation for hereditary neuropathies proceeds in stepwise fashion, beginning with the study of individual genes. If this fails to detect any abnormality, NGS analysis, which involves the sequencing of many different genes in parallel and has now become available for routine diagnosis, should be performed early on in the diagnostic work-up. Exome and genome analyses are currently performed only when considered to be indicated in the individual case. Whenever a hereditary neuropathy is suspected, other (including potentially treatable) causes of neuropathy should be ruled out. Mutations in neurop athy-associated genes may also be associated with other clinical entities such as spastic paraplegia or myopathy. Thus, interdisciplinary assessment is necessary.
Conclusion: The molecular diagnosis of neuropathies has become much more successful through the use of NGS. Although causally directed treatment approaches still need to be developed, the correct diagnosis puts an end to the often highly stressful search for a cause and enables determination of the risk of disease in other members of the patient's family.
Figures


References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical