

Biallelic Mutations in ATP5F1D, which Encodes a Subunit of ATP Synthase, Cause a Metabolic Disorder
- PMID: 29478781
- PMCID: PMC6117612
- DOI: 10.1016/j.ajhg.2018.01.020
Biallelic Mutations in ATP5F1D, which Encodes a Subunit of ATP Synthase, Cause a Metabolic Disorder
Abstract
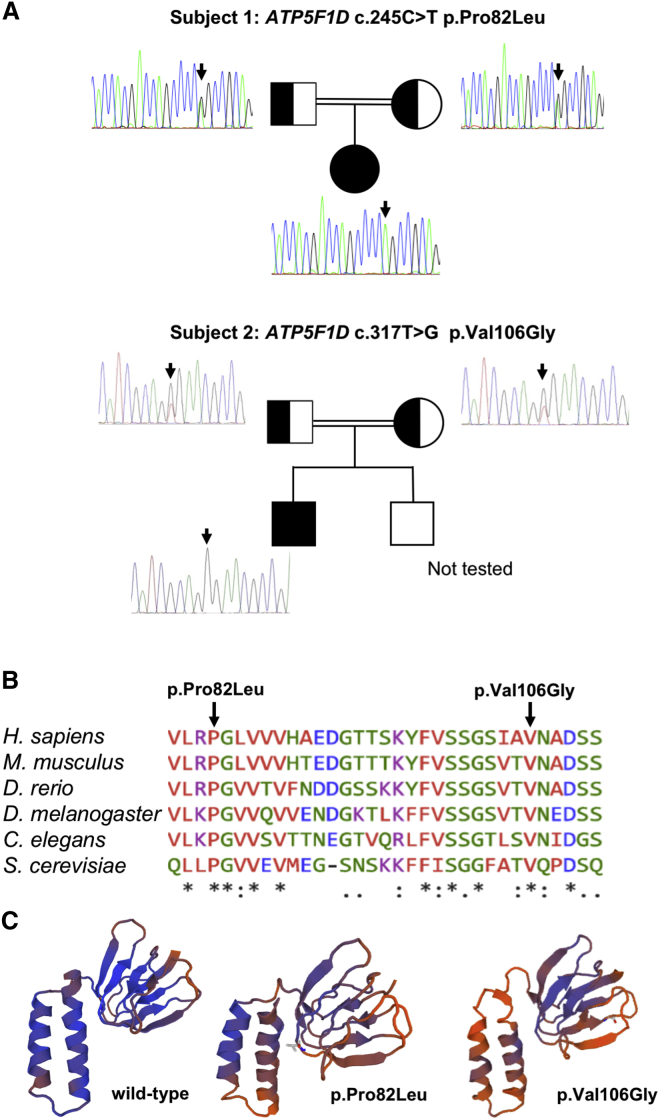
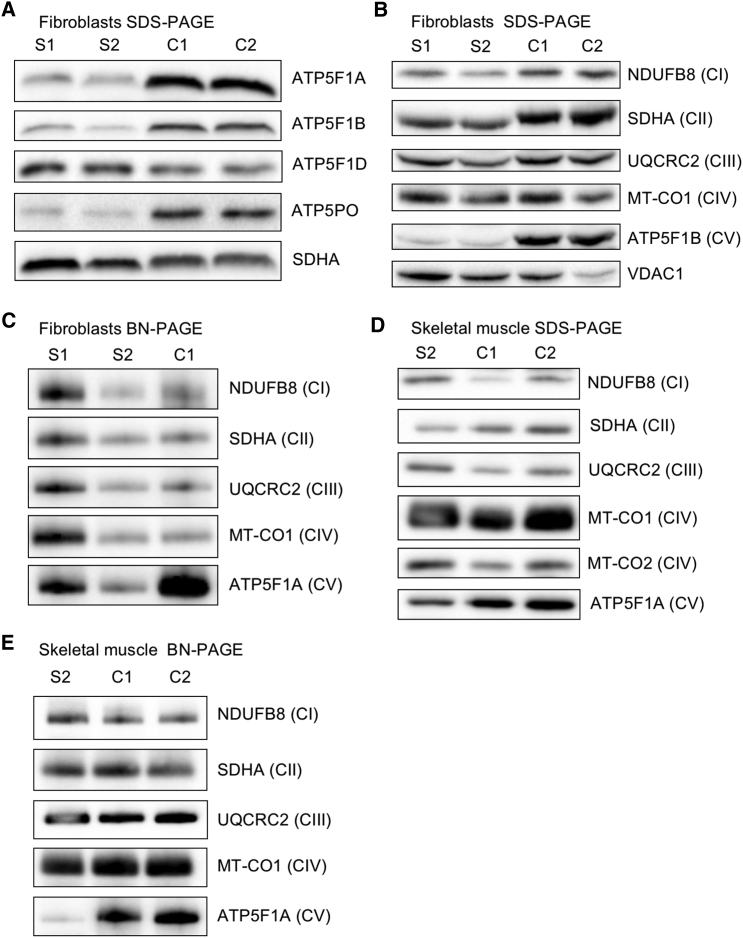
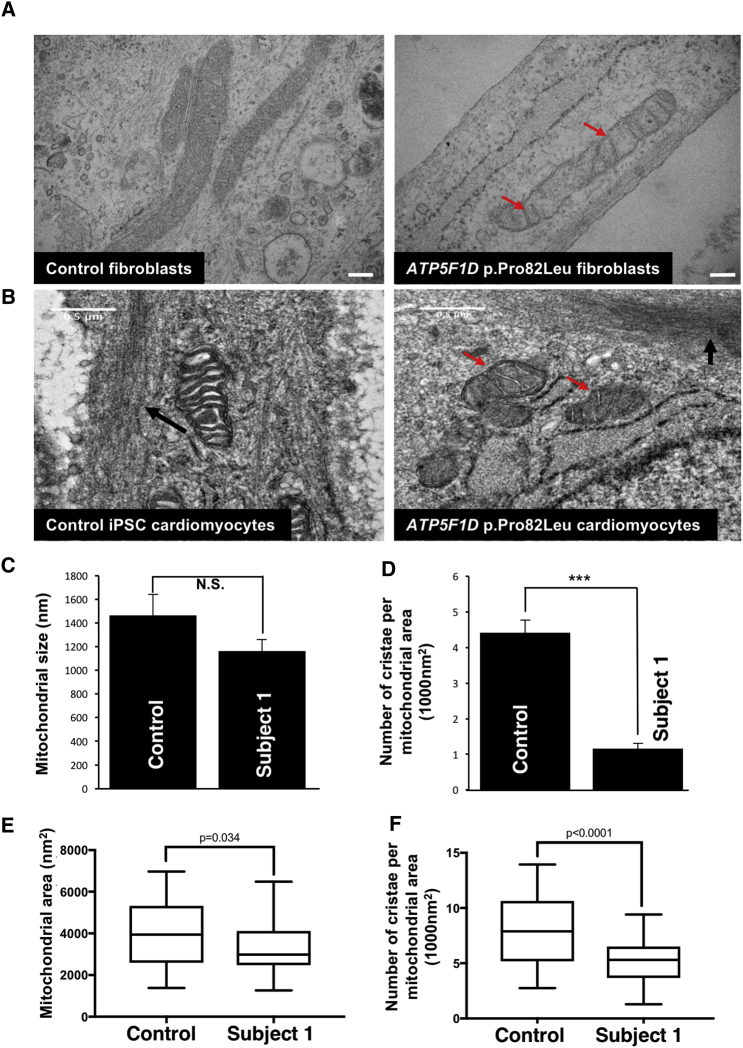
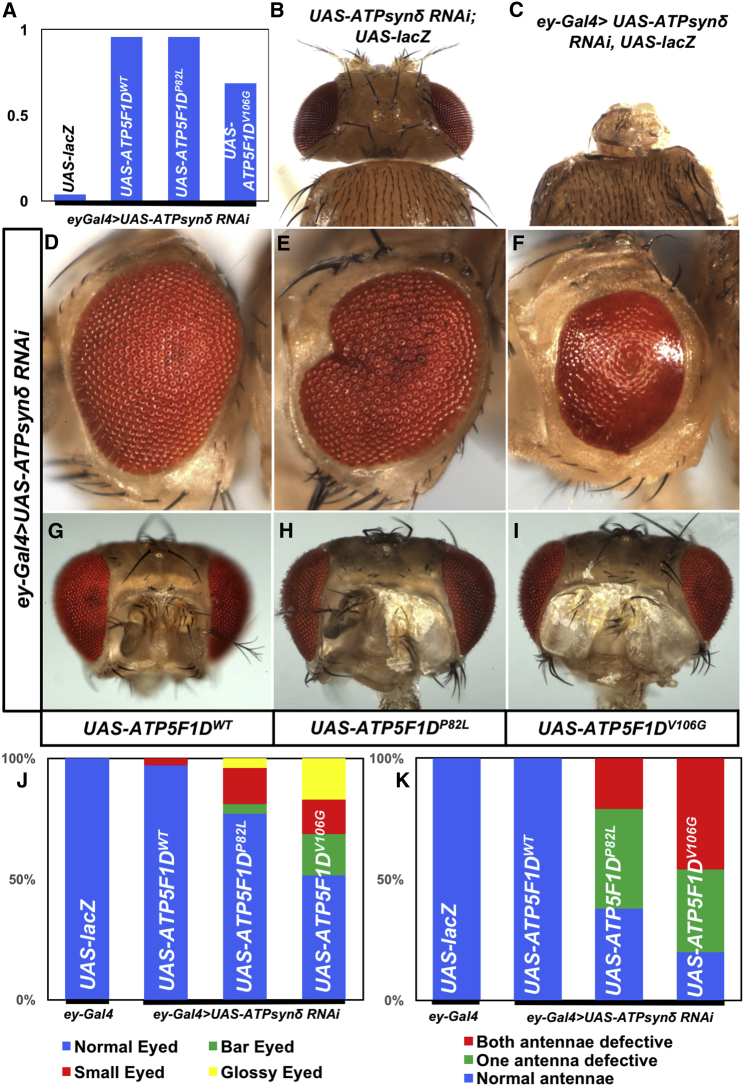
ATP synthase, H+ transporting, mitochondrial F1 complex, δ subunit (ATP5F1D; formerly ATP5D) is a subunit of mitochondrial ATP synthase and plays an important role in coupling proton translocation and ATP production. Here, we describe two individuals, each with homozygous missense variants in ATP5F1D, who presented with episodic lethargy, metabolic acidosis, 3-methylglutaconic aciduria, and hyperammonemia. Subject 1, homozygous for c.245C>T (p.Pro82Leu), presented with recurrent metabolic decompensation starting in the neonatal period, and subject 2, homozygous for c.317T>G (p.Val106Gly), presented with acute encephalopathy in childhood. Cultured skin fibroblasts from these individuals exhibited impaired assembly of F1FO ATP synthase and subsequent reduced complex V activity. Cells from subject 1 also exhibited a significant decrease in mitochondrial cristae. Knockdown of Drosophila ATPsynδ, the ATP5F1D homolog, in developing eyes and brains caused a near complete loss of the fly head, a phenotype that was fully rescued by wild-type human ATP5F1D. In contrast, expression of the ATP5F1D c.245C>T and c.317T>G variants rescued the head-size phenotype but recapitulated the eye and antennae defects seen in other genetic models of mitochondrial oxidative phosphorylation deficiency. Our data establish c.245C>T (p.Pro82Leu) and c.317T>G (p.Val106Gly) in ATP5F1D as pathogenic variants leading to a Mendelian mitochondrial disease featuring episodic metabolic decompensation.
Keywords: 3-methylglutaric aciduria; ATP synthase; complex V; exome sequencing; fibroblast; hyperammonemia; lactic acidosis; mitochondrial disease; model organism; oxidative phosphorylation.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
