

PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells
- PMID: 29479065
- PMCID: PMC5943191
- DOI: 10.1038/s41375-017-0008-6
PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells
Abstract
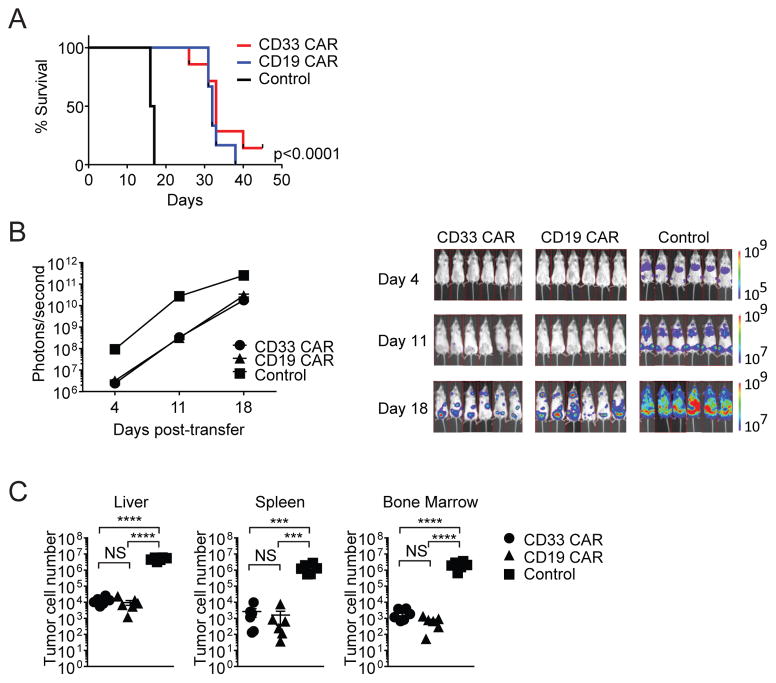
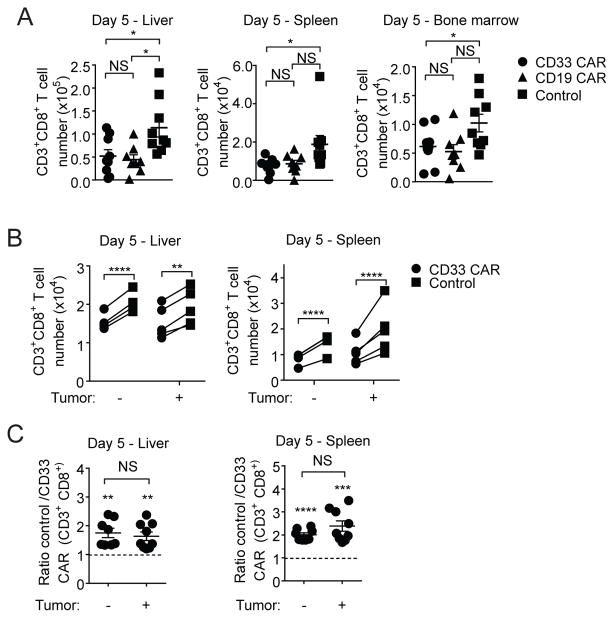
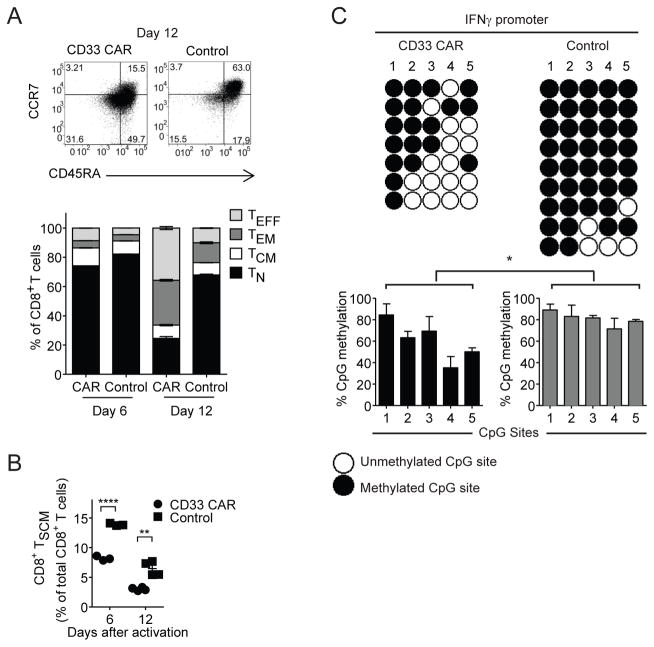
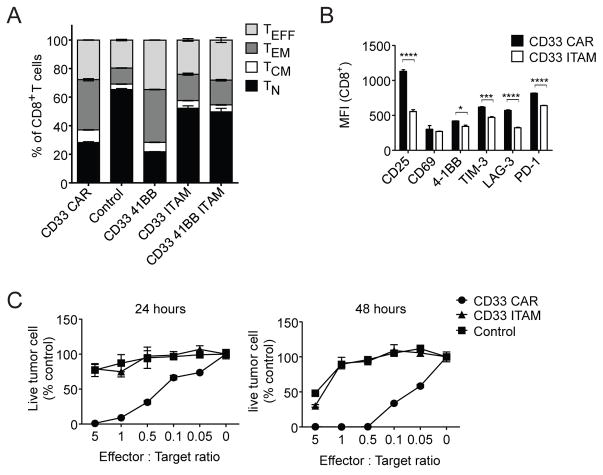
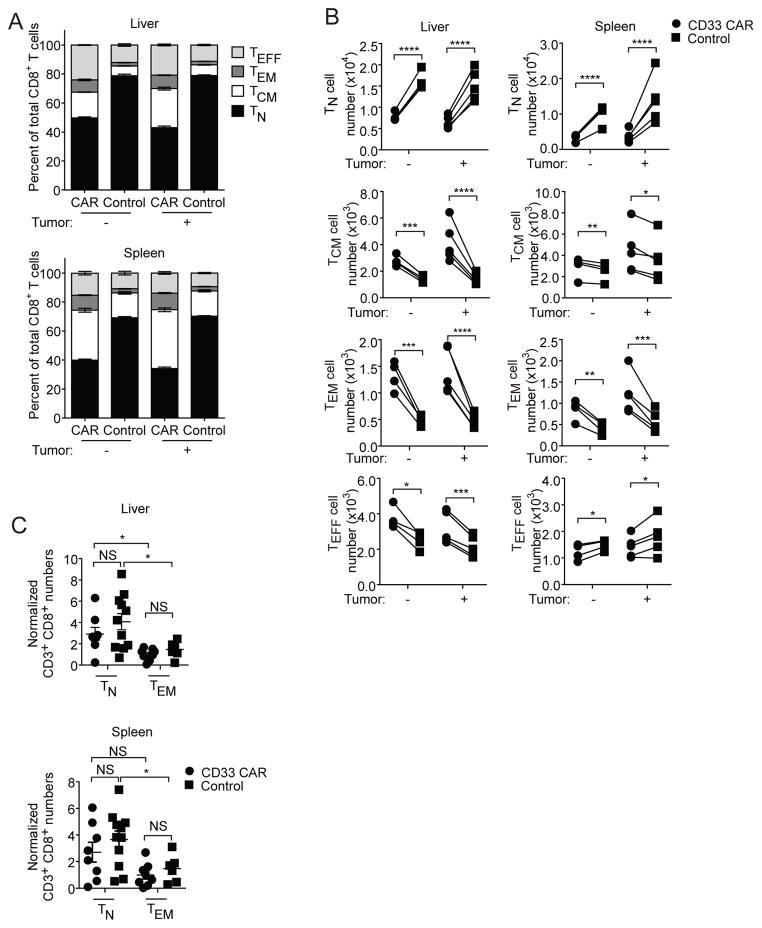
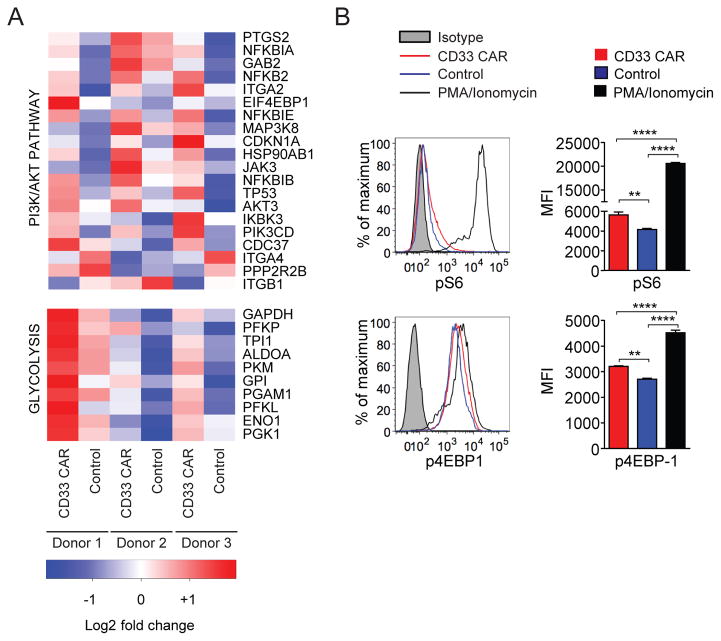
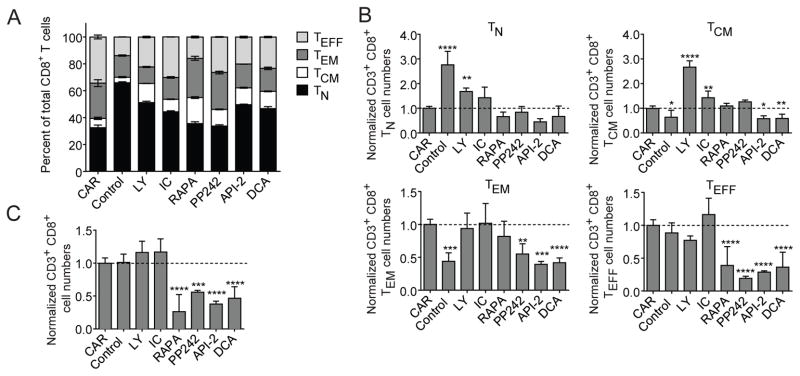
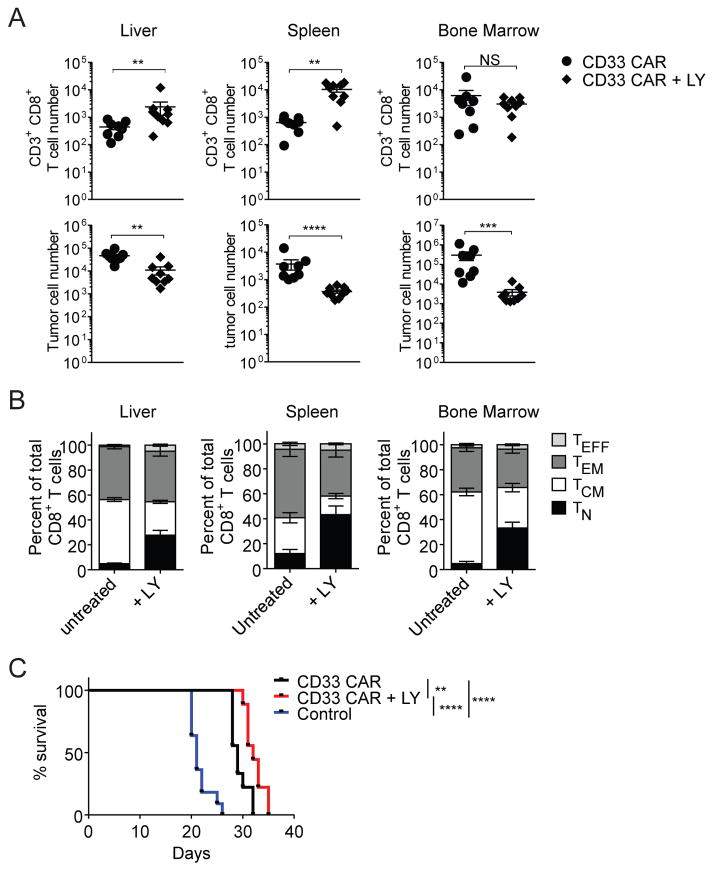
In vivo persistence of chimeric antigen receptor (CAR)-modified T cells correlates with therapeutic efficacy, yet CAR-specific factors that support persistence are not well resolved. Using a CD33-specific CAR in an acute myeloid leukemia (AML) model, we show how CAR expression alters T cell differentiation in a ligand independent manner. Ex vivo expanded CAR-T cells demonstrated decreased naïve and stem memory populations and increased effector subsets relative to vector-transduced control cells. This was associated with reduced in vivo persistence. Decreased persistence was not due to specificity or tumor presence, but to pre-transfer tonic signaling through the CAR CD3ζ ITAMs. We identified activation of the PI3K pathway in CD33 CAR-T cells as responsible. Treatment with a PI3K inhibitor modulated the differentiation program of CAR-T cells, preserved a less differentiated state without affecting T cell expansion, and improved in vivo persistence and reduced tumor burden. These results resolve mechanisms by which tonic signaling of CAR-T cells modulates their fate, and identifies a novel pharmacologic approach to enhance the durability of CAR-T cells for immunotherapy.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Figures
References
-
- Casucci M, Nicolis di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013 Nov 14;122(20):3461–3472. - PubMed
-
- Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, Spinelli O, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014 Aug;28(8):1596–1605. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
