

Review
doi: 10.1038/s41536-018-0045-4.
eCollection 2018.
Humanizing the mdx mouse model of DMD: the long and the short of it
Affiliations
- PMID: 29479480
- PMCID: PMC5816599
- DOI: 10.1038/s41536-018-0045-4
Item in Clipboard
Review
Humanizing the mdx mouse model of DMD: the long and the short of it
NPJ Regen Med.
.
Erratum in
-
Author Correction: Humanizing the mdx mouse model of DMD: the long and the short of it.NPJ Regen Med. 2020 Dec 21;5(1):25. doi: 10.1038/s41536-020-00112-0. NPJ Regen Med. 2020. PMID: 33349636 Free PMC article. No abstract available.
Abstract
Duchenne muscular dystrophy (DMD) is a common fatal heritable myopathy, with cardiorespiratory failure occurring by the third decade of life. There is no specific treatment for DMD cardiomyopathy, in large part due to a lack of understanding of the mechanisms underlying the cardiac failure. Mdx mice, which have the same dystrophin mutation as human patients, are of limited use, as they do not develop early dilated cardiomyopathy as seen in patients. Here we summarize the usefulness of the various commonly used DMD mouse models, highlight a model with shortened telomeres like humans, and identify directions that warrant further investigation.
Conflict of interest statement
The authors declare no competing interests.
Figures
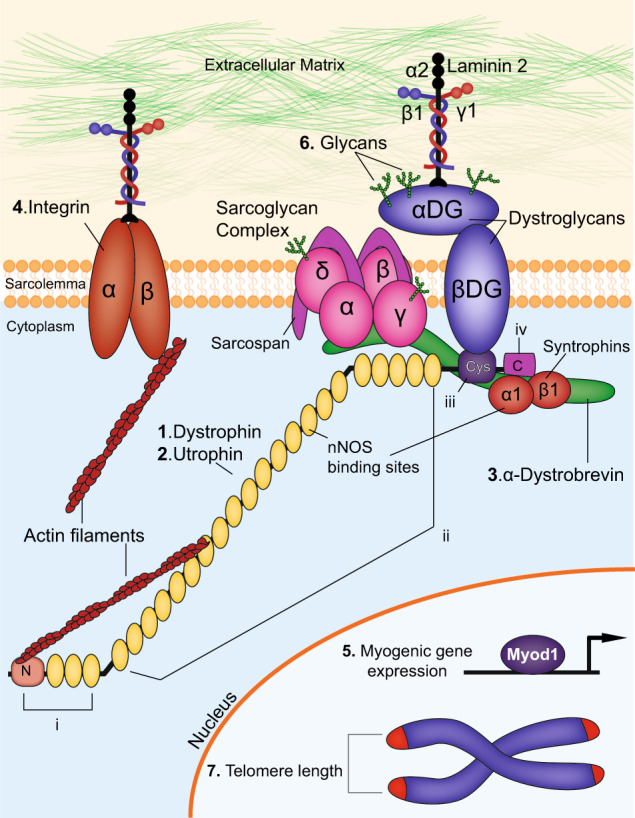
Components of the dystrophin-associated glycoprotein complex. The dystrophin-associated glycoprotein complex (DGC) and proteins that interact with DGC are depicted here. Extracellular, membrane, cytoplasmic and nuclear components are shown. The proteins targeted in double knockout mouse models of DMD, as outlined in the text, are indicated: (1) Dystrophin (2) Utrophin (3) α-Dystrobrevin (4) α7-Integrin (5) Myogenic differentiation factor 1(Myod) (6) Glycans and (7) telomere length. The domains of dystrophin are also indicated: (i) N-terminal domain (ii) middle body domain, which includes the nNOS binding sites (iii) cysteine-rich domain and (iv) C-terminal domain. In addition, DMD disease modifiers not directly involved in the DGC but that are also targeted in double knockout studies are also highlighted
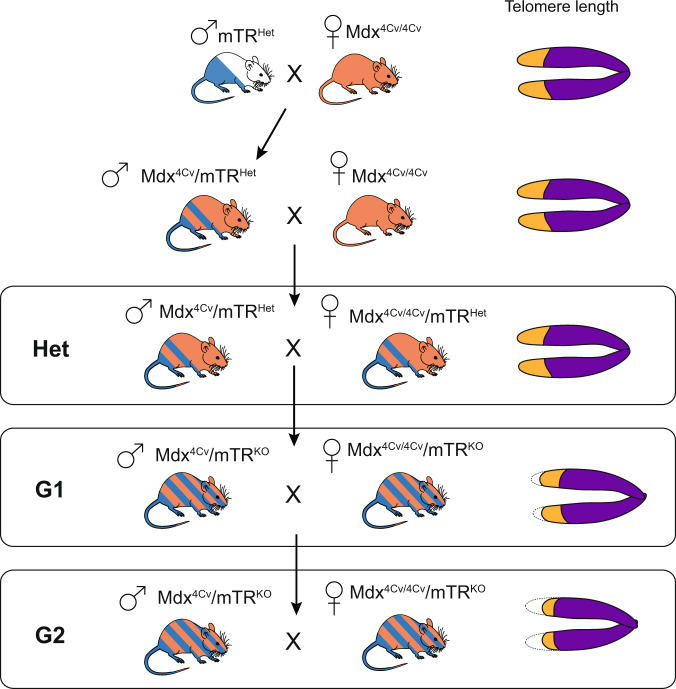
Crossing scheme for generation of the mdx4cv/mTRG2 model of Duchenne Muscular Dystrophy. Breeding begins with an initial cross of a male mTRHet heterozygous animal with a female mdx4cv/4cv homozygous animal, both of which are available live. The male progeny of this cross are bred with female mdx4cv/4cv animals. It should be noted that because the dystrophin gene is X-linked, males are inherently hemizygous while females are homozygous. Progeny of the mdx4cv/mTRHet x mdx4cv/4cv cross are denoted as “Het”. Male Het animals are heterozygous for the mTR mutation and have the mdx4cv mutation on the X-chromosome. Given the one functional copy of Terc (mTR), Hets with normal telomere lengths are used as controls. To create the first generation of mdx4cv/mTRKO (designated as mdx4cv/mTRG1) animals with shortened telomeres, inter-cousin breeding of mdx4cv/mTRHet is performed to avoid genetic drift., Finally, mdx4cv/mTRG1 animals are crossed, again through inter-cousin breeding, to generate the second generation mdx4cv/mTRG2 animals. mdx4cv/mTRG2 animals have “humanized” telomere lengths that allow the full penetrance of skeletal and cardiac muscle phenotypes
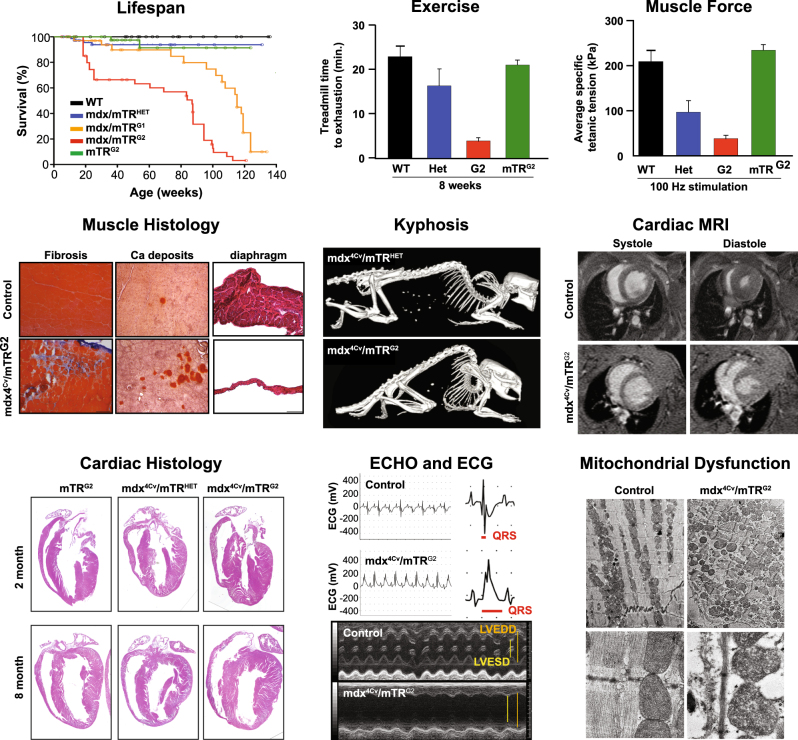
mdx4cv/mTRG2 animal model faithfully recapitulates the skeletal and the cardiac phenotype of DMD. Molecular and functional characterization of skeletal muscle and cardiac phenotypes in the mdx4cv/mTRG2 double knockout mouse. Overall, mdx4cv/mTRG2 exhibited decreased life span. In the skeletal muscle, mdx4cv/mTRG2 exhibited decreased exercise capacity, decreased muscle strength, histological evidence of muscular dystrophy, and kyphosis. In the heart, mdx4cv/mTRG2 displayed cardiac dysfunction as measured by MRI, histology, ECHO, ECG, and electron microscopy. Figure adapted from prior mdx4cv/mTRG2 studies,
References
-
- Monaco AP, et al. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323:646. - PubMed
-
- Hoffman EP, Kunkel LM. Dystrophin abnormalities in Duchenne/Becker muscular dystrophy. Neuron. 1989;2:1019–1029. - PubMed
-
- Emery AEH. The muscular dystrophies. Lancet. 2002;359:687–695. - PubMed
-
- Chyatte S, Vignos PJ, Watkins M. Early muscular dystrophy: differential patterns of weakness in Duchenne, limb-girdle and facioscapulohumeral types. Arch. Phys. Med. Rehabil. 1966;47:499–503. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
