

Genetics of Autosomal Recessive Polycystic Kidney Disease and Its Differential Diagnoses
- PMID: 29479522
- PMCID: PMC5811498
- DOI: 10.3389/fped.2017.00221
Genetics of Autosomal Recessive Polycystic Kidney Disease and Its Differential Diagnoses
Abstract
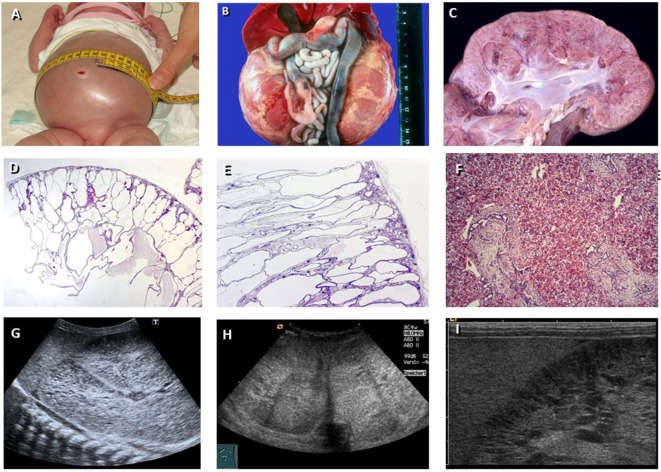
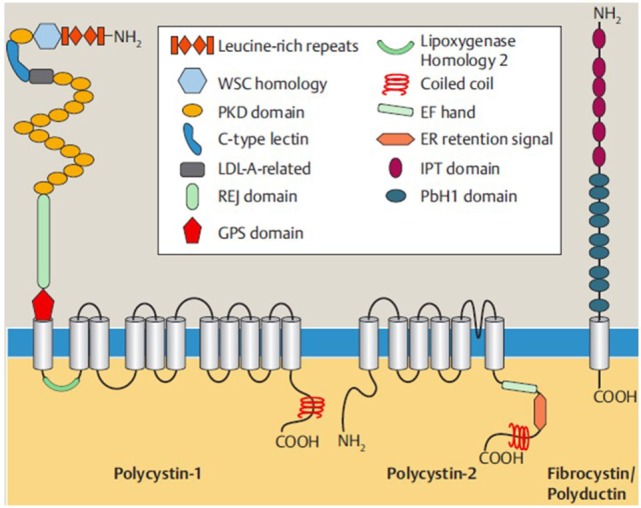
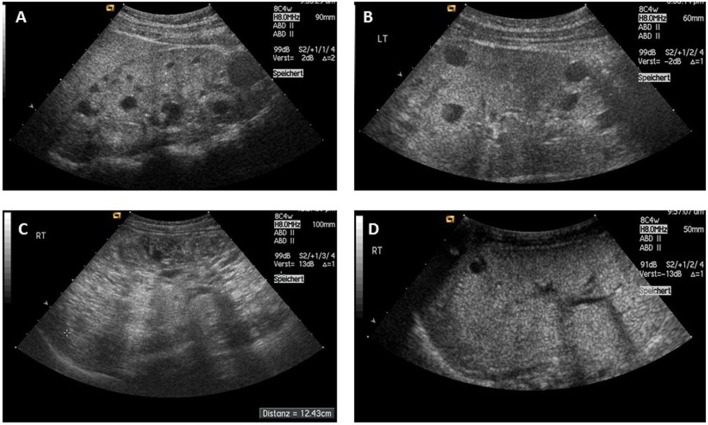
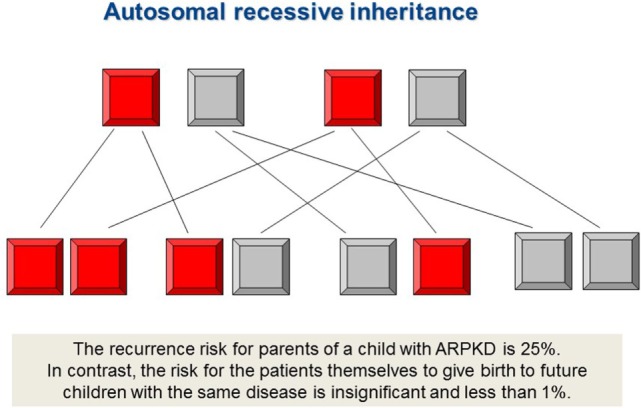
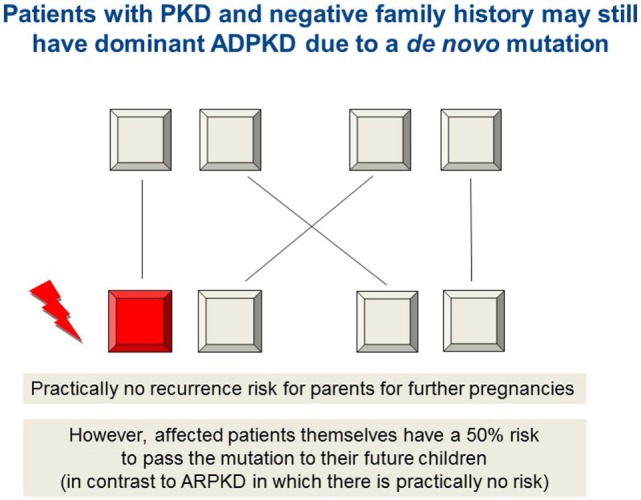
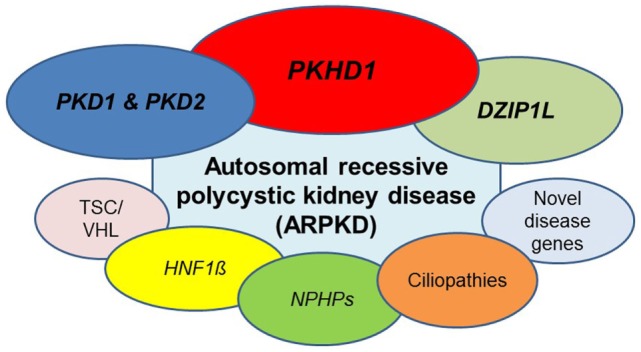
Autosomal recessive polycystic kidney disease (ARPKD) is a hepatorenal fibrocystic disorder that is characterized by enlarged kidneys with progressive loss of renal function and biliary duct dilatation and congenital hepatic fibrosis that leads to portal hypertension in some patients. Mutations in the PKHD1 gene are the primary cause of ARPKD; however, the disease is genetically not as homogeneous as long thought and mutations in several other cystogenes can phenocopy ARPKD. The family history usually is negative, both for recessive, but also often for dominant disease genes due to de novo arisen mutations or recessive inheritance of variants in genes that usually follow dominant patterns such as the main ADPKD genes PKD1 and PKD2. Considerable progress has been made in the understanding of polycystic kidney disease (PKD). A reduced dosage of disease proteins leads to the disruption of signaling pathways underlying key mechanisms involved in cellular homeostasis, which may help to explain the accelerated and severe clinical progression of disease course in some PKD patients. A comprehensive knowledge of disease-causing genes is essential for counseling and to avoid genetic misdiagnosis, which is particularly important in the prenatal setting (e.g., preimplantation genetic diagnosis/PGD). For ARPKD, there is a strong demand for early and reliable prenatal diagnosis, which is only feasible by molecular genetic analysis. A clear genetic diagnosis is helpful for many families and improves the clinical management of patients. Unnecessary and invasive measures can be avoided and renal and extrarenal comorbidities early be detected in the clinical course. The increasing number of genes that have to be considered benefit from the advances of next-generation sequencing (NGS) which allows simultaneous analysis of a large group of genes in a single test at relatively low cost and has become the mainstay for genetic diagnosis. The broad phenotypic and genetic heterogeneity of cystic and polycystic kidney diseases make NGS a particularly powerful approach for these indications. Interpretation of genetic data becomes the challenge and requires deep clinical understanding.
Keywords: ADPKD; DZIP1L; HNF1β/TCF2; PKD1/PKD2; PKHD1; autosomal recessive polycystic kidney disease (ARPKD); ciliopathies; nephronophthisis (NPHP).
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous