

17-AAG inhibits vemurafenib-associated MAP kinase activation and is synergistic with cellular immunotherapy in a murine melanoma model
- PMID: 29481571
- PMCID: PMC5826531
- DOI: 10.1371/journal.pone.0191264
17-AAG inhibits vemurafenib-associated MAP kinase activation and is synergistic with cellular immunotherapy in a murine melanoma model
Abstract
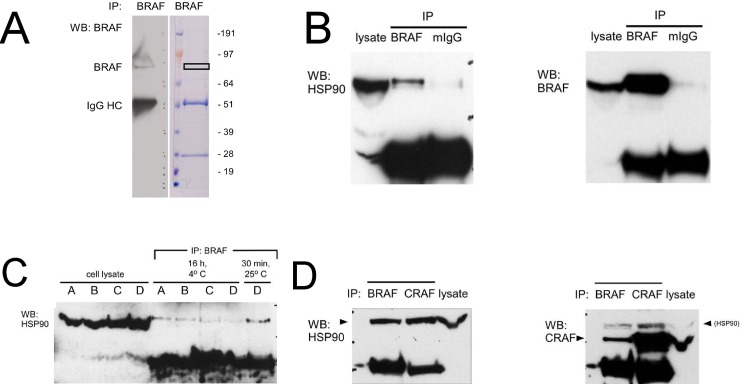
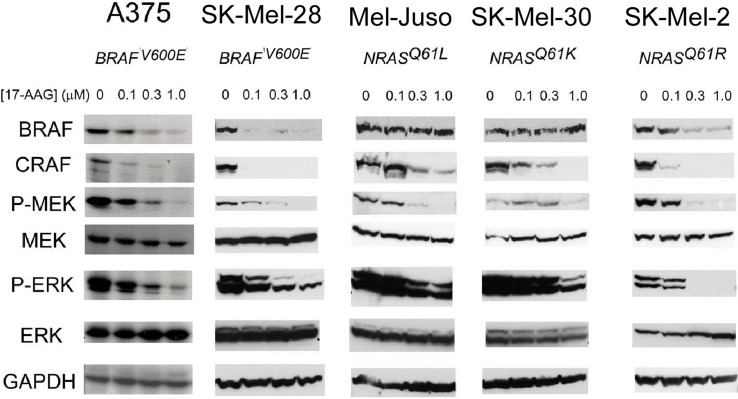
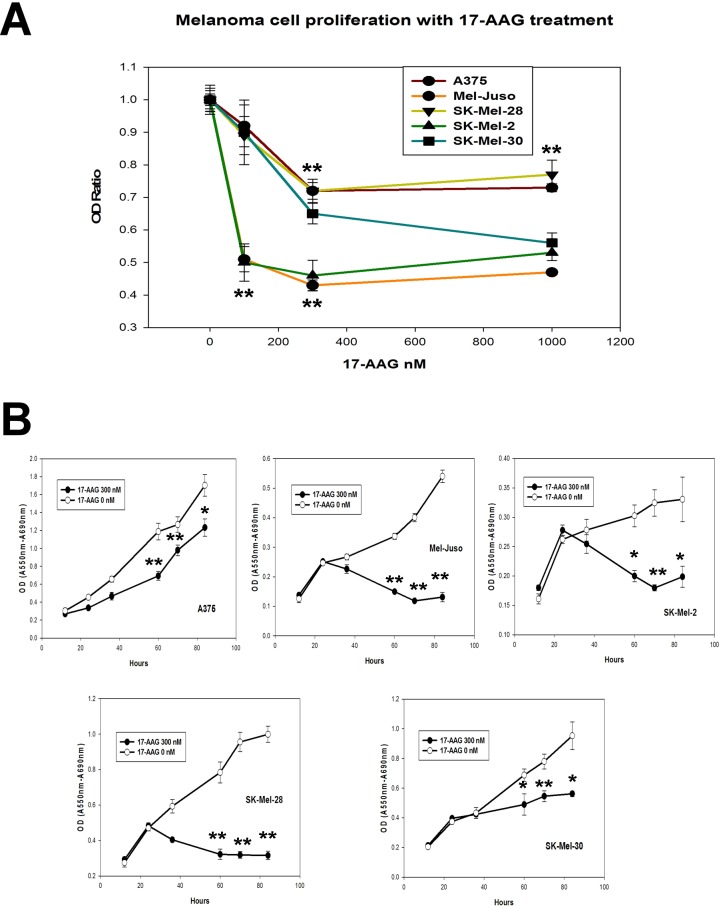
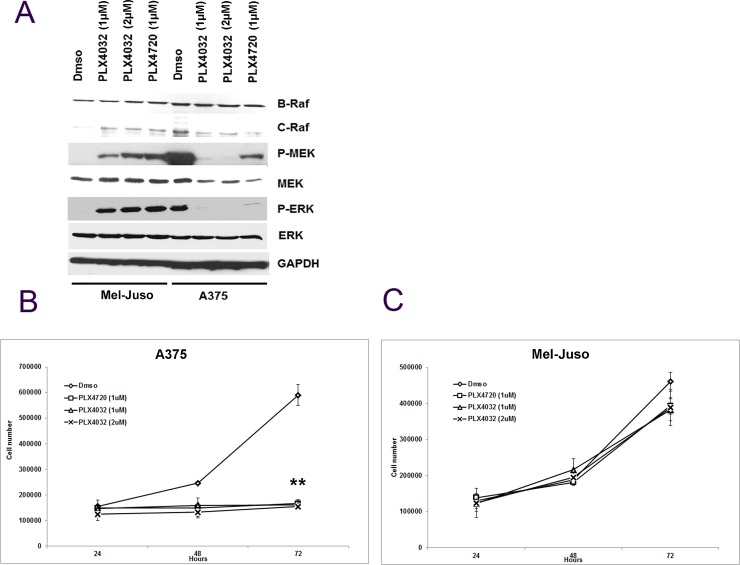
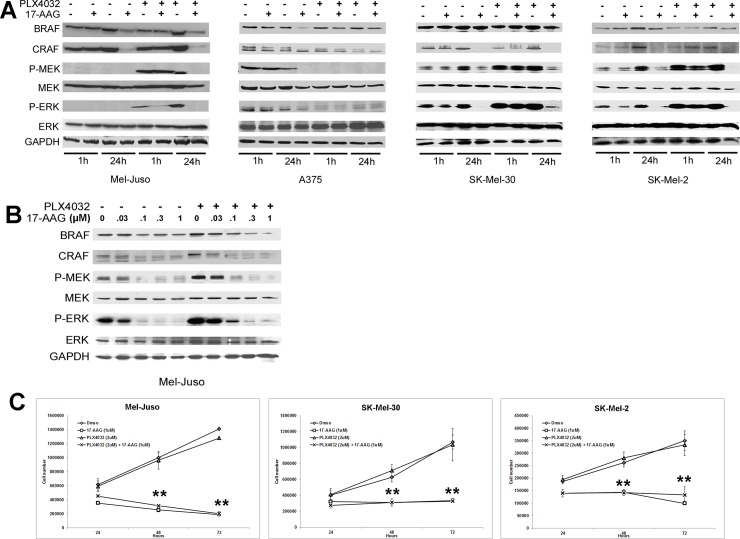
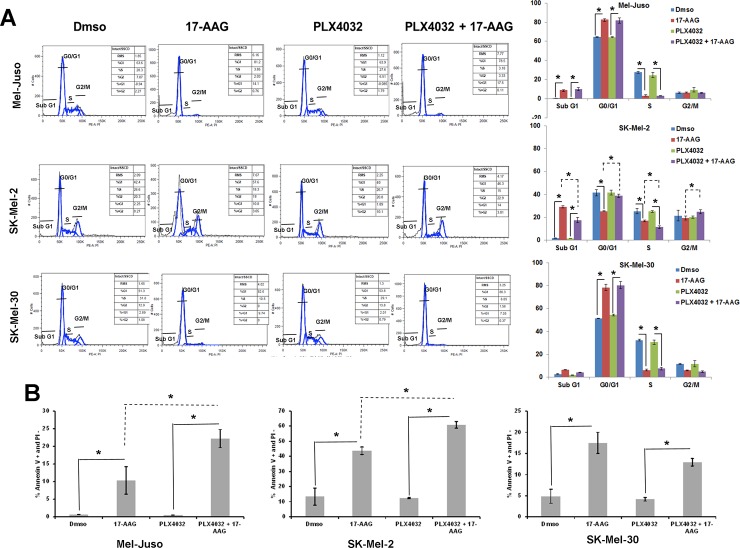
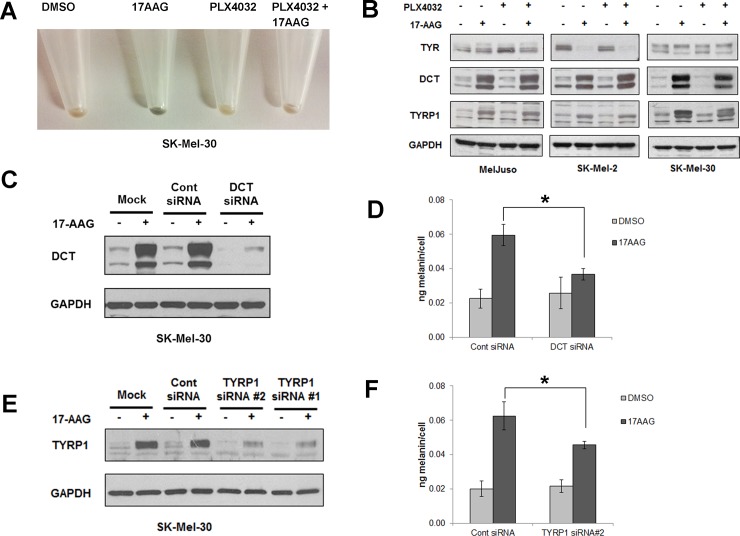
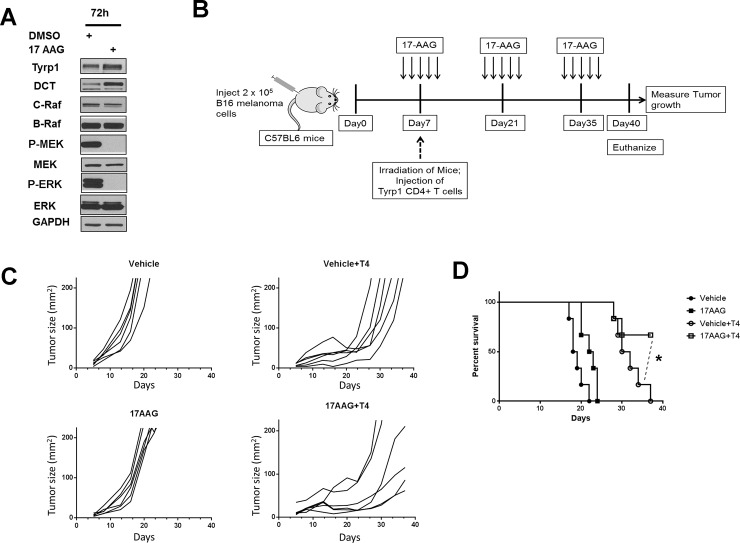
Heat shock protein 90 (HSP90) is a molecular chaperone which stabilizes client proteins with important roles in tumor growth. 17-allylamino-17-demethoxygeldanamycin (17-AAG), an inhibitor of HSP90 ATPase activity, occupies the ATP binding site of HSP90 causing a conformational change which destabilizes client proteins and directs them towards proteosomal degradation. Malignant melanomas have active RAF-MEK-ERK signaling which can occur either through an activating mutation in BRAF (BRAFV600E) or through activation of signal transduction upstream of BRAF. Prior work showed that 17-AAG inhibits cell growth in BRAFV600E and BRAF wildtype (BRAFWT) melanomas, although there were conflicting reports about the dependence of BRAFV600E and BRAFWT upon HSP90 activity for stability. Here, we demonstrate that BRAFWT and CRAF are bound by HSP90 in BRAFWT, NRAS mutant melanoma cells. HSP90 inhibition by 17-AAG inhibits ERK signaling and cell growth by destabilizing CRAF but not BRAFWT in the majority of NRAS mutant melanoma cells. The highly-selective BRAFV600E inhibitor, PLX4032 (vemurafenib), inhibits ERK signaling and cell growth in mutant BRAF melanoma cells, but paradoxically enhances signaling in cells with wild-type BRAF. In our study, we examined whether 17-AAG could inhibit PLX4032-enhanced ERK signaling in BRAFWT melanoma cells. As expected, PLX4032 alone enhanced ERK signaling in the BRAFWT melanoma cell lines Mel-Juso, SK-Mel-2, and SK-Mel-30, and inhibited signaling and cell growth in BRAFV600E A375 cells. However, HSP90 inhibition by 17-AAG inhibited PLX4032-enhanced ERK signaling and inhibited cell growth by destabilizing CRAF. Surprisingly, 17-AAG also stimulated melanin production in SK-Mel-30 cells and enhanced TYRP1 and DCT expression without stimulating TYR production in all three BRAFWT cell lines studied as well as in B16F10 mouse melanoma cells. In vivo, the combination of 17-AAG and cellular immunotherapy directed against Tyrp1 enhanced the inhibition of tumor growth compared to either therapy alone. Our studies support a role for 17-AAG and HSP90 inhibition in enhancing cellular immunotherapy for melanoma.
Conflict of interest statement
Figures
References
-
- Neckers L. (2007) Heat shock protein 90: The cancer chaperone. J Biosci 32(3): 517–530. - PubMed
-
- Xu W, Mimnaugh E, Rosser MF, Nicchitta C, Marcu M, et al. (2001) Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J Biol Chem 276(5): 3702–3708. doi: 10.1074/jbc.M006864200 - DOI - PubMed
-
- Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, et al. (2002) Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem 277(42): 39858–39866. doi: 10.1074/jbc.M206322200 - DOI - PubMed
-
- Schulte TW, Blagosklonny MV, Ingui C, Neckers L. (1995) Disruption of the raf-1-Hsp90 molecular complex results in destabilization of raf-1 and loss of raf-1-ras association. J Biol Chem 270(41): 24585–24588. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
